Author: Denis Avetisyan
Researchers have developed a new framework to dissect the inner workings of protein language models, revealing the computational circuits that drive biological function.
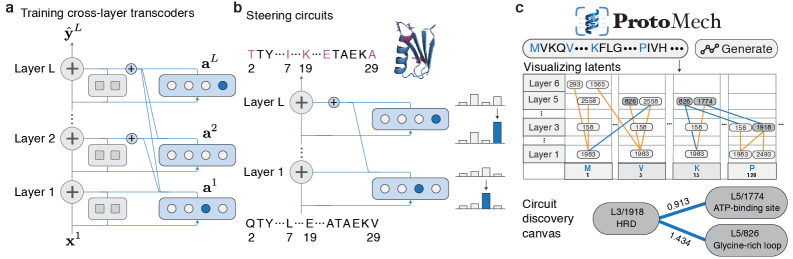
ProtoMech leverages cross-layer transcoders and sparse autoencoders to discover and analyze circuits within protein language models, achieving state-of-the-art performance in mechanistic interpretability.
Despite the success of protein language models (pLMs) in predicting protein structure and function, their internal computational logic remains largely opaque. To address this, we present ‘Protein Circuit Tracing via Cross-layer Transcoders’, introducing ProtoMech, a framework that leverages cross-layer transcoders to discover interpretable circuits within pLMs. This approach recovers 82-89% of original performance while identifying compressed circuits-using <1% of latent space-that correlate with key structural and functional motifs and even enable improved protein design. Can these discovered circuits ultimately unlock a deeper understanding of protein biology and accelerate the development of novel therapeutics?
Decoding Protein Intelligence: Unveiling the Hidden Logic
Protein Language Models (PLMs), such as ESM2, have recently achieved unprecedented success in predicting protein structure and function from amino acid sequences. These models, trained on vast datasets of protein sequences, exhibit a remarkable ability to discern patterns and relationships previously inaccessible to traditional computational methods. However, this proficiency comes at a cost: the internal workings of PLMs remain largely opaque. Despite their predictive power, the specific mechanisms by which these models arrive at their conclusions are poorly understood, effectively functioning as ‘black boxes’. While a PLM can accurately predict a protein’s properties, deciphering how it arrived at that prediction – which residues or patterns were most influential – presents a significant challenge. This lack of transparency limits the potential for rational protein design, where scientists aim to engineer proteins with specific, desired characteristics, and hinders the development of truly interpretable artificial intelligence in the biological realm.
The promise of protein language models extends far beyond mere sequence completion; realizing their full potential hinges on deciphering the computational processes occurring within these complex systems. A thorough understanding of how PLMs internally represent and process information is not simply an academic pursuit, but a prerequisite for rational protein design – the ability to engineer proteins with specific, predetermined functions. Without insight into these internal computations, manipulating PLMs remains largely trial-and-error. Furthermore, accurately predicting protein function – a critical bottleneck in biological research – depends on interpreting the model’s reasoning, not just its output. By unlocking the ‘black box’ of PLMs, researchers can move beyond correlation to causation, enabling the design of novel proteins tailored to address challenges in medicine, materials science, and beyond, and ultimately accelerating the pace of biological discovery.
Despite the impressive performance of Protein Language Models (PLMs) in predicting protein structure and function, a significant challenge lies in deciphering how these models arrive at their conclusions. Current analytical techniques often fail to pinpoint the specific, modular computations within the PLM that govern its behavior – essentially treating the model as a complex, opaque system. This lack of interpretability severely limits the ability to rationally design proteins with desired characteristics or reliably predict the functional consequences of specific mutations. Without understanding these internal ‘circuits’, researchers are constrained to empirical approaches, hindering the full exploitation of PLMs’ predictive power and slowing the progress of protein engineering and drug discovery. Identifying these discrete, interpretable components is therefore critical for transitioning from observation to true understanding and control over protein intelligence.

ProtoMech: A Framework for Dissecting Computational Logic
ProtoMech employs Cross-Layer Transcoders (CLTs) as the primary mechanism for dimensionality reduction of hidden states within a Pre-trained Language Model (PLM). These transcoders function by projecting the high-dimensional hidden state representation at each layer of the PLM into a lower-dimensional Latent Space. This mapping is achieved through learned linear transformations, effectively compressing the information contained within each layer’s hidden state while preserving salient features for subsequent analysis and reconstruction. The resulting Latent Space representation serves as a condensed proxy for the original hidden state, enabling efficient computation and improved interpretability of the PLM’s internal workings.
Cross-Layer Transcoders (CLTs) function by establishing a learned mapping between the input and output states of each layer within a Product Lifecycle Management (PLM) model. This is achieved through training individual transcoders to approximate the transformation performed by each layer, effectively capturing the directional flow of information. By analyzing these input-output relationships at each stage, the CLTs build a representation of how data propagates through the network, allowing for reconstruction of the original PLM’s computational path. This layer-wise mapping provides granular insight into the network’s processing of information and forms the basis for a simplified, interpretable model.
The Replacement Model functions as a distilled representation of the original Product Lifecycle Management (PLM) model’s computational process. Constructed using the Cross-Layer Transcoders (CLTs) – which map hidden states to a lower-dimensional Latent Space – this model offers a significantly reduced parameter count compared to the full PLM. This simplification facilitates improved interpretability; by isolating and representing the core information flow through the CLTs, the Replacement Model allows for analysis of individual layers and their contributions to the overall computation. The resulting approximation maintains key functionalities while providing a more tractable and understandable system for examining PLM behavior.

Validating Computational Models: Evidence from Experiment
Circuit validation utilizes supervised learning techniques to determine the predictive power of discovered circuits regarding protein family membership. This process involves training a classifier on circuit activity – typically represented by the values of latent variables – and evaluating its ability to correctly assign proteins to their respective families. Performance is quantified using standard classification metrics, such as accuracy, precision, and recall. Successful prediction of protein family membership indicates that the identified circuit captures functionally relevant information and accurately reflects the underlying biological computation governing protein behavior. This approach provides a quantitative assessment of circuit validity and allows for comparison between different circuit discovery methods.
Deep Mutational Scanning (DMS) assays are employed to quantitatively assess the functional impact of amino acid mutations on protein fitness. These assays involve creating comprehensive libraries of protein variants, each containing a single amino acid mutation, and measuring their relative abundance or activity under selective conditions. The resulting data establishes a quantitative relationship between genotype (mutational profile) and phenotype (fitness or function). By correlating experimentally derived fitness effects with the predicted activity of identified circuits, we can validate the model’s ability to accurately represent the biological consequences of genetic variation and establish a link between circuit behavior and observed phenotypic effects.
ProtoMech demonstrates high performance in recovering original model capabilities, achieving 89% accuracy in protein family classification and an 82% Spearman correlation for function prediction. This level of performance is attained through the identification of minimal sets of latent variables that effectively govern model behavior, enabling a reductionist approach to understanding complex protein computations. By mapping these variables, ProtoMech provides a simplified representation of the underlying mechanisms driving protein function, allowing for focused analysis and improved predictive power.
Functional Steering: Precision Engineering with Latent Space Control
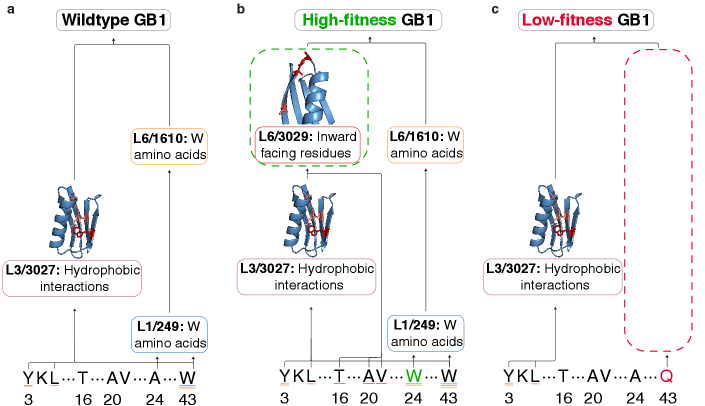
ProtoMech facilitates what researchers term “Functional Steering” – the ability to predictably alter a protein’s characteristics – by operating within the protein’s ‘Latent Space’. This space represents a compressed, high-dimensional map of all possible protein structures and functions. Instead of directly modifying amino acid sequences, ProtoMech identifies and manipulates specific ‘circuits’ – interconnected patterns of activity – within this Latent Space. By subtly adjusting these circuits, the system can effectively ‘steer’ the protein towards a desired function, such as enhanced binding affinity or altered catalytic activity. This approach bypasses the need for exhaustive trial-and-error methods traditionally used in protein engineering, offering a far more targeted and efficient pathway to designing proteins with tailored properties.
The architecture of complex protein function is often obscured within the vastness of sequence space, but ProtoMech introduces a method for revealing these underlying relationships through the visualization of ‘Virtual Weights’. These weights, representing the strength of connections between different points in the latent space, effectively map the flow of information within a protein’s functional circuitry. By rendering these connections, researchers can discern how specific regions of the latent space contribute to desired functional attributes, identifying key pathways and dependencies that govern protein behavior. This approach moves beyond simple correlation to reveal the causal architecture of function, allowing for targeted manipulation of these circuits to predictably engineer proteins with tailored properties – offering a level of precision previously unattainable in rational protein design.
The efficiency of ProtoMech in navigating the vast landscape of protein sequence space is remarkable, achieving performance on par with existing methods while utilizing a surprisingly small subset of parameters – a sparse circuit comprising just 96 latents, or approximately 0.8% of the total latent space. This streamlined approach not only reduces computational demands but also demonstrably surpasses the performance of PLT, a leading protein language model, in 71% of circuit steering scenarios. By effectively focusing on the most crucial elements within the latent space, ProtoMech unlocks a powerful strategy for generating protein sequences tailored to specific functional attributes, representing a significant step towards realizing the long-held goal of rational protein design and offering a potentially transformative tool for biotechnology and medicine.
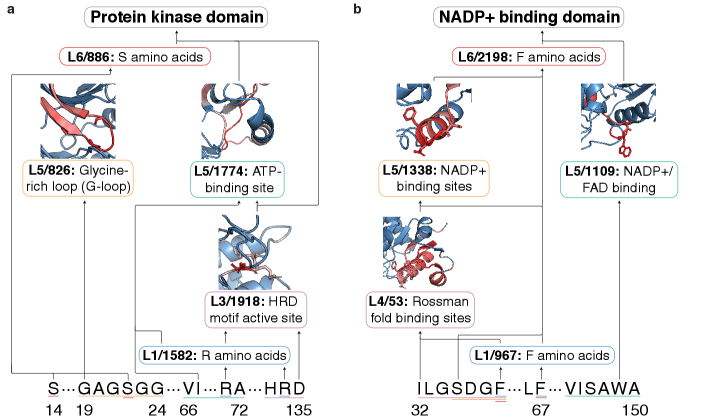
The pursuit of mechanistic interpretability, as demonstrated by ProtoMech’s cross-layer transcoders, echoes a fundamental principle of efficient communication. The framework distills complex protein language model behavior into sparse, identifiable circuits – a process akin to reducing noise and amplifying signal. Andrey Kolmogorov observed, “The errors are more important than the truths.” This resonates deeply with the work; identifying where a model deviates from expected behavior, through circuit tracing, is paramount. ProtoMech doesn’t merely confirm functionality, but dissects the underlying logic, acknowledging and addressing the ‘errors’ inherent in the system’s computation. The focus on sparsity further exemplifies this clarity, prioritizing essential information over superfluous detail.
What Remains to Be Seen?
The pursuit of mechanistic interpretability, as exemplified by this work, invariably reveals the limits of current decomposition. ProtoMech offers a compelling advance in circuit tracing, yet the recovered motifs, however biologically suggestive, remain abstractions. The true elegance of a system is not found in the complexity of its components, but in the parsimony of their interactions. Future work must address the inherent difficulty in establishing a ground truth – the living system is not a static circuit diagram, but a dynamic, noisy process.
The reliance on protein language models, while currently productive, introduces a potential for circularity. These models are trained on biological data; to interpret them as revealing fundamental principles risks mistaking correlation for causation. A fruitful avenue lies in developing methods that can directly interrogate the physical instantiation of these circuits – to move beyond the symbolic representation and grapple with the messy reality of molecular interactions.
Ultimately, the goal is not simply to map the circuitry, but to understand the why of it. Why these particular arrangements? What selective pressures have shaped these computational strategies? The answers, one suspects, will not be found in the details of the code, but in the history of life itself.
Original article: https://arxiv.org/pdf/2602.12026.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-15 02:57