Author: Denis Avetisyan
Researchers have developed a powerful new method to predict where proteins are located within tissues, even when direct protein measurements are limited.
![The STProtein training framework leverages a multi-stage approach-initial protein structure prediction, followed by iterative refinement using [latex] \nabla_{\theta} L(\theta, x) [/latex]-to sculpt protein conformations capable of fulfilling designated functional requirements, ultimately demonstrating an adaptive system responding to the inherent entropy of structural possibilities.](https://arxiv.org/html/2602.05811v1/images/structure/framework4.jpg)
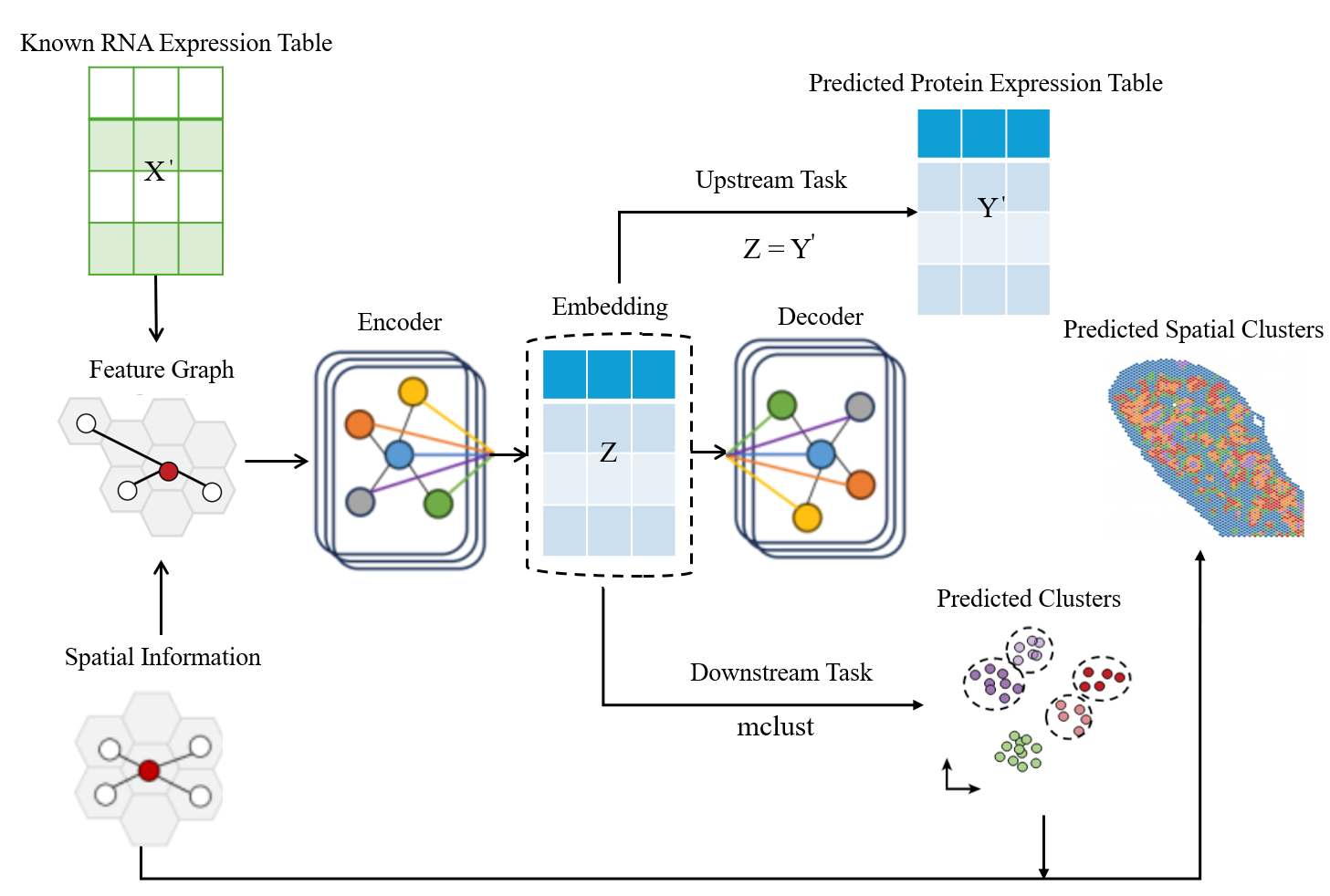
STProtein leverages graph neural networks to accurately predict spatial protein expression by integrating data from multiple omics sources.
Despite advances in spatial multi-omics, a significant data imbalance hinders comprehensive tissue characterization-spatial transcriptomics data is abundant, while spatial proteomics remains limited by cost and technical challenges. To address this, we present ‘STProtein: predicting spatial protein expression from multi-omics data’, a novel framework leveraging graph neural networks and multi-task learning to accurately predict spatial protein expression from more accessible multi-omics data. This approach effectively bridges the gap created by sparse spatial proteomics, enabling deeper insights into tissue biology and the exploration of previously hidden spatial patterns. Will STProtein unlock a new era of discovery by revealing the complex interplay between genes, proteins, and spatial organization within tissues?
The Erosion of Homogeneity: A Spatial Biology Imperative
Historically, biological investigations have frequently treated tissues as homogenous samples, averaging cellular properties and losing vital information about the intricate organization within. This approach overlooks the fact that cell behavior is profoundly influenced by its immediate surroundings – its neighbors, the extracellular matrix, and distance from blood vessels. Consequently, traditional methods, like bulk RNA sequencing, provide only an aggregate picture, obscuring the spatial relationships that govern processes like development, disease progression, and therapeutic response. The inability to resolve this spatial context has limited understanding of how cellular heterogeneity contributes to overall tissue function and has hindered the development of truly targeted therapies, emphasizing the need for techniques capable of mapping molecular data directly onto the tissue architecture.
The advent of spatial omics – technologies like spatial transcriptomics, proteomics, and metabolomics – is revolutionizing biological research by enabling the simultaneous measurement of multiple molecular features within the intricate context of tissue architecture. However, this powerful capability generates datasets of unprecedented scale and complexity, presenting a significant analytical challenge. Integrating these diverse layers of ‘omic’ data – gene expression, protein abundance, metabolic profiles, and spatial location – requires novel computational approaches and bioinformatics pipelines. Simply compiling the information isn’t enough; researchers must develop methods to effectively harmonize, analyze, and interpret these multi-dimensional datasets to reveal meaningful biological insights, a task that demands both increased computational power and innovative algorithms capable of extracting signal from the noise of such complex systems.
Biological data isn’t limited to the genes and proteins scientists actively study; a substantial portion remains unannotated, often referred to as ‘dark matter’. This unexplored territory within tissues and cells represents a significant frontier in biological research. Advanced spatial omics technologies are now capable of mapping the molecular profiles of these previously ignored regions, revealing unexpected patterns and potential regulatory elements. Deciphering this ‘dark matter’ isn’t simply about identifying unknown molecules; it’s about understanding how these uncharacterized areas contribute to cellular function, disease progression, and the overall complexity of biological systems. The ability to illuminate this hidden landscape promises to reshape our understanding of life itself, potentially uncovering novel biomarkers and therapeutic targets previously obscured by a lack of spatial resolution and comprehensive data analysis.
![STProtein accurately predicts gene expression in the mouse thymus [latex]CITE[/latex]-seq dataset, demonstrating performance comparable to established benchmarking methods when compared to original ground truth.](https://arxiv.org/html/2602.05811v1/images/clustering/Mouse_Thymus.png)
STProtein: Reconstructing the Molecular Landscape
STProtein is a computational framework developed for the prediction of spatially-resolved protein expression levels. The method integrates multi-omic data – including genomics, transcriptomics, and proteomics – to infer protein abundance at specific locations within a tissue sample. This is achieved by constructing a predictive model trained on the combined dataset, enabling the estimation of protein expression patterns directly from spatial coordinates and associated omic profiles. The framework aims to provide insights into protein localization and function within the context of the tissue microenvironment, facilitating a more comprehensive understanding of biological processes.
STProtein employs a K-Nearest Neighbors (KNN) feature graph to represent spatial relationships within tissue samples. This graph construction method identifies the ‘k’ most similar spatial locations based on shared multi-omic feature vectors – including gene expression, protein abundance, and morphological characteristics – and establishes edges connecting these locations. The resulting KNN graph effectively captures local tissue structure by prioritizing proximity in feature space as a proxy for physical adjacency. The value of ‘k’ is a user-defined parameter influencing the granularity of the captured relationships; lower values emphasize immediate neighbors, while higher values incorporate broader contextual information. This graph-based representation allows the model to propagate information between spatially-correlated locations, enhancing prediction accuracy by leveraging the inherent organization of the tissue.
STProtein employs Graph Neural Networks (GNNs) to analyze spatial transcriptomic data by representing tissue architecture as a graph where nodes represent spatial locations and edges define relationships between them. GNNs operate directly on this graph structure, enabling the model to learn node embeddings that capture both local and global spatial context. This approach allows STProtein to effectively integrate information from neighboring locations, identifying complex patterns in gene expression that would be difficult to detect using traditional methods. The network-based analysis facilitates the prediction of protein expression levels at each spatial location by propagating information across the graph and leveraging learned relationships between genes, proteins, and spatial context.

Validation and the Measure of Predictive Fidelity
Root Mean Squared Error (RMSE) was utilized as the primary metric for evaluating the predictive accuracy of STProtein against other benchmarking methods. Comparative analysis across multiple datasets revealed STProtein consistently outperforms alternatives; specifically, STProtein achieved an RMSE that is 0.04 lower on Mouse Spleen, 0.07 lower on Mouse Thymus, and 0.17 lower on Human Lymph Node datasets. These results demonstrate STProtein’s enhanced capability in accurately predicting spatial transcriptomics data compared to existing approaches, as lower RMSE values indicate reduced prediction error.
STProtein utilizes the Seurat toolkit for data preprocessing, a widely adopted approach in single-cell spatial transcriptomics analysis. This includes standard quality control procedures, normalization, and feature selection. To manage the high dimensionality inherent in spatial transcriptomics data and facilitate efficient computation, the framework incorporates dimensionality reduction techniques such as Principal Component Analysis (PCA) and Uniform Manifold Approximation and Projection (UMAP). PCA identifies principal components that capture the maximum variance in the data, while UMAP focuses on preserving the global structure of the data in a lower-dimensional space, enabling effective visualization and downstream analysis.
STProtein incorporates clustering methodologies that consistently outperform comparative methods across a suite of validation metrics. Specifically, the framework achieves the highest values for Normalized Mutual Information (NMI), Adjusted Mutual Information (AMI), Fowlkes-Mallows Index (FMI), Adjusted Rand Index (ARI), Homogeneity, V-measure, and F1-Score. These metrics assess the quality of identified clusters based on both internal consistency and external validation against known ground truth, indicating STProtein’s superior ability to accurately group and categorize spatial transcriptomic data.
Toward a Predictive Histology: Implications and Future Trajectories
A significant hurdle in spatial multi-omics analysis lies in data imbalance – certain cell types or molecular features are often far less represented than others, potentially skewing analytical results. STProtein directly confronts this challenge through a novel statistical framework designed to appropriately weight contributions from all data points, regardless of their frequency. By mitigating the influence of dominant signals, the method achieves more robust and reliable predictions of cellular states and spatial patterns. This balanced approach is crucial for accurately characterizing complex tissues and identifying subtle but important biological differences, ultimately enhancing the interpretability and translational potential of spatial omics data.
The predictive capabilities of STProtein extend beyond descriptive analysis, offering substantial promise for translational applications in biomedicine. By accurately deconvoluting spatial transcriptomics data, the method facilitates a deeper understanding of disease progression at the cellular level, enabling researchers to pinpoint critical molecular changes associated with various stages of illness. This detailed insight can then be leveraged to identify potential therapeutic targets – specific molecules or pathways that, when modulated, could halt or reverse disease development. Furthermore, STProtein’s ability to characterize cellular heterogeneity within tissues opens avenues for personalized treatment strategies, tailoring interventions based on an individual’s unique molecular profile and the specific characteristics of their disease manifestation. Ultimately, this approach aims to move beyond generalized treatments towards precision medicine, maximizing efficacy and minimizing adverse effects.
Continued development of STProtein centers on a multi-pronged approach to refine its predictive capabilities and biological insights. Researchers plan to incorporate data from additional omics layers – such as proteomics and metabolomics – to create a more holistic view of spatial biology and improve prediction accuracy. Simultaneously, the integration of transfer learning techniques, including scArches and totalVI, promises to leverage existing single-cell data to overcome limitations imposed by sparse spatial datasets. Notably, the application of STProtein to mouse spleen tissue yielded the identification and characterization of two previously unannotated macrophage subsets, designated MZMΦ and MMMΦ, significantly enriching the current understanding of splenic composition and potentially revealing novel roles for these cells in immune function and disease pathogenesis.
![STProtein accurately predicts gene expression in human lymph node tissue [latex]10x[/latex] Genomics Visium data, demonstrating improved performance compared to benchmarking prediction methods.](https://arxiv.org/html/2602.05811v1/images/clustering/Human_Lymph_Node.png)
The pursuit of accurately modeling biological systems, as demonstrated by STProtein’s framework for predicting spatial protein expression, echoes a fundamental truth about all complex structures. Like infrastructure slowly succumbing to the forces of entropy, biological data-even at its most granular level-is inherently subject to incompleteness. STProtein attempts to bridge the gap created by limited spatial proteomics data, effectively patching against inevitable decay. As Donald Knuth observed, “Premature optimization is the root of all evil,” and this work embodies that wisdom; rather than striving for unattainable perfection, the framework focuses on practical prediction from available multi-omics data, accepting inherent limitations while striving for graceful aging of the model’s predictive power. This approach ensures continued utility even as the underlying biological landscape shifts and evolves.
What’s Next?
The prediction of spatial protein expression from multi-omics data, as demonstrated by STProtein, represents a temporary caching of a fundamental scarcity. The true limitation isn’t algorithmic-graph neural networks will inevitably refine-but the inherent latency in acquiring comprehensive spatial proteomics. Each predicted protein is, after all, a request paid for with the cost of incomplete observation. The framework itself functions as a bridge, extending insight beyond the currently measurable, but the gulf between transcript and protein, between prediction and reality, remains.
Future iterations will undoubtedly focus on expanding the modalities integrated within these predictive models. However, the persistent challenge lies in acknowledging the system’s eventual decay. Noise accumulates, biological contexts shift, and the correlation between transcriptome and proteome will inevitably degrade over time. Uptime, in this sense, is merely a statistical anomaly.
The true metric isn’t accuracy, but graceful degradation. A resilient framework will not attempt to eliminate error, but to model it, incorporating uncertainty as a first-class citizen. The goal isn’t a perfect map, but a reliable estimate of the territory’s inevitable change, accepting that all systems are, ultimately, transient flows.
Original article: https://arxiv.org/pdf/2602.05811.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- The Division Resurgence Best Weapon Guide: Tier List, Gear Breakdown, and Farming Guide
- Kagurabachi Chapter 118 Release Date, Time & Where to Read Manga
- Last Furry: Survival redeem codes and how to use them (April 2026)
- Clash of Clans Sound of Clash Event for April 2026: Details, How to Progress, Rewards and more
- Guild of Monster Girls redeem codes and how to use them (April 2026)
- GearPaw Defenders redeem codes and how to use them (April 2026)
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- All Mobile Games (Android and iOS) releasing in April 2026
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
- Limbus Company 2026 Roadmap Revealed
2026-02-08 04:15