Author: Denis Avetisyan
Researchers have developed a new AI framework that accurately predicts the arrangement of molecules in organic crystals, a critical step for designing advanced materials.
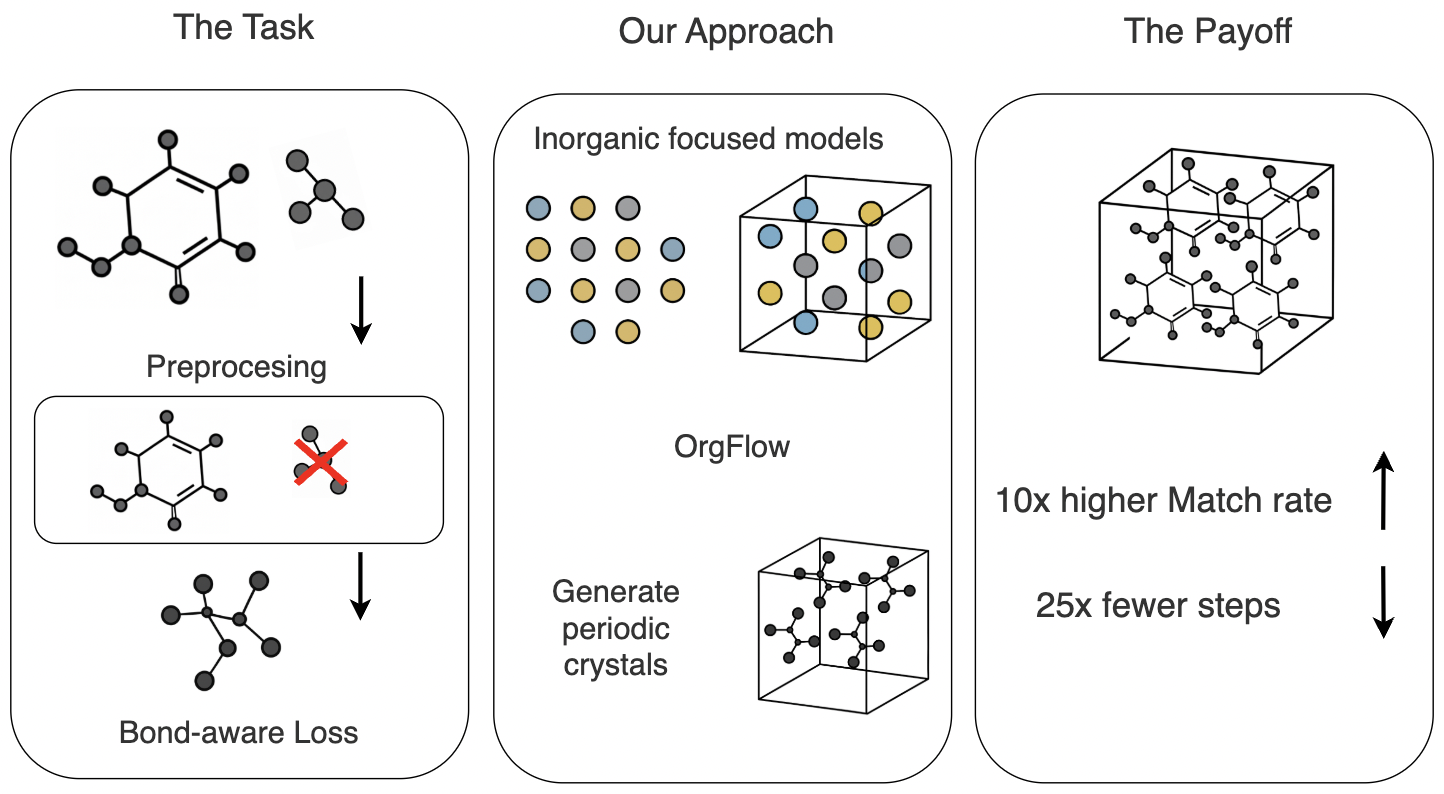
OrgFlow utilizes flow matching and molecular graph conditioning to surpass existing crystal structure prediction methods, particularly for organic compounds.
Predicting crystal structures remains a significant challenge in materials science, with existing data-driven approaches largely focused on inorganic compounds. This limitation motivates the development of new methods tailored to organic crystals-essential components of pharmaceuticals, polymers, and functional materials-which present unique complexities due to larger unit cells and strict chemical connectivity. Here, we introduce ‘OrgFlow: Generative Modeling of Organic Crystal Structures from Molecular Graphs’, a novel framework leveraging a flow-matching network conditioned on molecular graphs and guided by a bond-aware loss to accurately and efficiently predict organic crystal structures. Does this approach pave the way for accelerated materials discovery and design in organic chemistry and beyond?
The Emergence of Order: Predicting Crystal Landscapes
The accurate determination of atomic arrangements within crystalline solids presents a persistent hurdle in materials science, with far-reaching implications for technological advancement. Predicting these stable configurations is not merely an academic exercise; it directly influences the efficacy of pharmaceuticals – where crystal structure dictates drug solubility and bioavailability – and the performance of energy storage devices, such as batteries, where ion transport is critically dependent on the material’s arrangement. Consequently, researchers are continually striving to refine computational methods capable of navigating the immense complexity of potential crystal structures, aiming to accelerate the discovery of novel materials with tailored properties and functionalities. The challenge lies in the exponential increase in computational demand as molecular complexity rises, necessitating innovative algorithms and high-performance computing to reliably forecast stable crystal packing arrangements.
Predicting the stable crystal structures of organic molecules presents a formidable computational challenge due to their inherent complexity and the sheer size of the chemical space they occupy. Unlike simpler inorganic compounds, organic molecules possess a multitude of degrees of freedom – rotations around single bonds, conformational changes, and diverse intermolecular interactions – that dramatically increase the search space for potential packing arrangements. Exhaustively exploring all possible configurations is computationally prohibitive, even with powerful supercomputers, as the number of possibilities grows exponentially with the number of atoms. This necessitates the development of sophisticated algorithms and force fields capable of efficiently navigating this vast landscape and accurately estimating the relative energies of different crystal structures, a task that remains a central focus of materials science research.
The accurate prediction of crystal structures hinges on a detailed understanding of how molecules interact and change shape within a crystal lattice. Intermolecular interactions – encompassing hydrogen bonds, van der Waals forces, and electrostatic effects – dictate the overall stability of a packing arrangement, yet are notoriously difficult to model with complete precision. Equally important is accounting for conformational flexibility; molecules aren’t rigid bodies, and their ability to rotate, vibrate, and adopt different shapes significantly influences how they pack together. Failing to accurately capture these dynamic aspects often leads to incorrect predictions, as even subtle changes in molecular conformation can dramatically alter the energy landscape and favor alternative crystal structures. Consequently, computational methods are continually refined to better represent these subtle, yet critical, factors, pushing the boundaries of what’s predictable in the realm of materials design.

Flow Matching: A Path Through Complexity
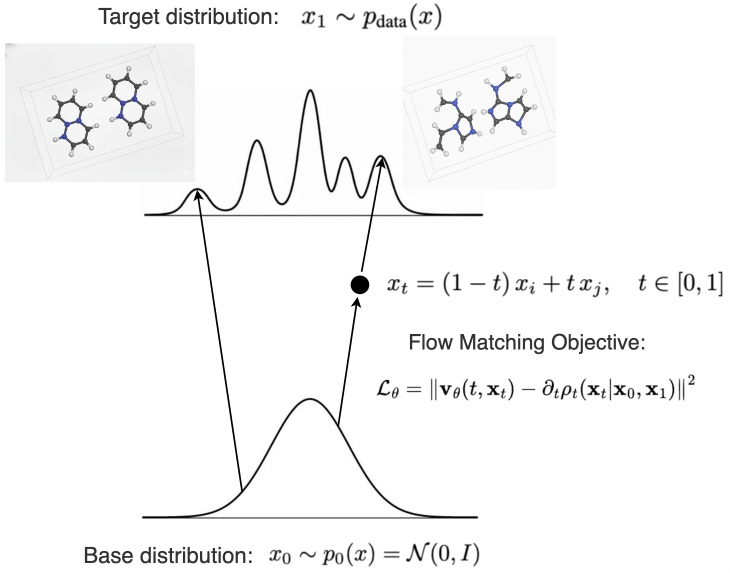
Flow Matching generates crystalline structures by learning a continuous, smooth transformation between probability distributions representing different configurations. This is achieved by defining a vector field that guides the generation process from a simple, known distribution to the complex distribution of target crystalline structures. The technique parameterizes the transformation as a time-dependent process, enabling the model to gradually evolve an initial random state into a realistic crystal conformation. By optimizing the vector field to match the gradient of the data distribution, Flow Matching effectively learns the underlying manifold of possible crystalline structures, allowing for the sampling of diverse and high-quality results. This approach differs from methods relying on discrete steps or Markovian assumptions by enabling direct, continuous generation without the limitations inherent in those models.
Traditional diffusion models rely on Markovian assumptions, requiring each step in the generative process to depend only on the previous state; this can introduce limitations in modeling complex data distributions and increase computational cost. Flow Matching circumvents this requirement by learning a continuous flow that directly maps data distributions, eliminating the need to define a Markov chain. This non-Markovian approach allows for potentially faster sampling, as fewer steps may be required to generate a sample, and improved stability during training, as the model is less susceptible to error accumulation inherent in iterative Markovian processes. The removal of Markovian constraints enables the model to learn more direct and efficient paths through the data space, ultimately improving both the speed and reliability of the generative process.
Defining a continuous flow in flow matching enables efficient exploration of the conformational landscape of molecules by representing the generation process as a trajectory through a series of increasingly realistic structures. This approach contrasts with discrete sampling methods and allows the model to navigate complex energy landscapes more effectively, avoiding local minima that might trap traditional optimization algorithms. By smoothly transforming a simple initial distribution into the target distribution of crystal structures, flow matching facilitates the discovery of novel crystal forms that would be difficult to identify using conventional methods reliant on stochastic search or limited conformational sampling. The continuous nature of the flow also provides gradients for optimization, allowing for directed exploration of the conformational space and accelerating the discovery process.
OrgFlow: A Chemically Informed Framework
OrgFlow employs a conditional flow-matching framework designed to generate geometrically valid crystal structures by explicitly representing molecular connectivity. This is achieved through the use of E(3)-Equivariant Message Passing, a technique that ensures predictions respect the rotational and translational symmetries inherent in 3D space. Message passing allows information about atoms and bonds to be iteratively updated, propagating structural information throughout the lattice. By incorporating these equivariant properties into the model architecture, OrgFlow avoids generating physically implausible configurations and maintains structural integrity during the generation process, critical for accurate crystal structure prediction.
OrgFlow incorporates a Bond-Aware Regularization Loss function during the generative modeling process to enforce chemically plausible structures. This loss term penalizes deviations from established bond length distributions observed in known molecular structures. Specifically, the loss function calculates the difference between predicted interatomic distances and statistically derived distributions of bond lengths for specific atom types. By minimizing this difference, the framework encourages the generation of molecules with realistic bond geometries, enhancing the chemical validity of the predicted crystal structures and improving the overall accuracy of the prediction process. This regularization is crucial for guiding the sampling process towards stable and physically meaningful configurations.
The implementation of Periodic Boundary Conditions (PBCs) within OrgFlow addresses a fundamental challenge in crystal structure prediction: accurately representing the infinite, repeating nature of crystal lattices. Without PBCs, simulations are limited by finite simulation box dimensions, introducing artificial constraints and potentially leading to inaccurate predictions of unit cell parameters and atomic positions. PBCs treat the simulation box as one unit within an infinitely extended lattice, effectively eliminating boundary effects by replicating the simulation box in all directions. When an atom crosses a boundary, its periodic image enters from the opposite side, maintaining a consistent density and avoiding spurious interactions caused by truncated long-range forces. This methodology ensures that predicted structures are representative of the bulk material and not artifacts of the simulation environment.
OrgFlow demonstrates a match rate of up to 13.6% when predicting organic crystal structures. This metric represents the percentage of generated structures that are considered valid matches to reference structures within a defined dataset. Importantly, this performance significantly exceeds that of existing crystal structure prediction methods, which were primarily designed and optimized for inorganic materials. These comparative methods typically exhibit substantially lower match rates when applied to the more complex and diverse chemical space of organic crystals, highlighting OrgFlow’s specific advantages in this domain. The reported 13.6% match rate indicates a notable improvement in the ability to accurately predict organic crystal structures compared to previous state-of-the-art approaches.
OrgFlow demonstrates a substantial improvement in predicting the correct crystal structures of drug-like molecules, achieving a match rate of 21.94%. This represents a significant advancement over baseline methods, which currently achieve a match rate of only 0.1% on the same datasets. The observed performance increase indicates that OrgFlow’s chemically aware framework effectively navigates the complex conformational space of drug-like compounds, leading to a significantly higher probability of generating accurate crystal structures.
OrgFlow significantly reduces the computational cost of crystal structure prediction by requiring 25 times fewer sampling steps than existing methods. This efficiency gain is achieved through the framework’s chemically aware design and conditional flow-matching approach, which constrains the search space to more plausible crystal configurations. Fewer sampling steps directly translate to reduced computational resources and faster prediction times, enabling the exploration of a larger chemical space and accelerating materials discovery efforts. The reduction in steps does not compromise accuracy, as demonstrated by the framework’s performance on both organic and drug-like molecules.

The Ripple Effect: Implications for Materials Discovery
The existence of multiple crystalline forms, known as polymorphism, profoundly impacts a material’s properties and, consequently, its performance in real-world applications. A single compound can exhibit drastically different solubility, stability, melting points, and even optical characteristics depending on its crystal packing. This phenomenon is particularly critical in pharmaceuticals, where polymorphism can affect drug bioavailability and efficacy – a different crystal form might dissolve too slowly or not at all, rendering a medication ineffective. Beyond drugs, controlling polymorphism is vital in designing high-performance organic electronics, where crystal structure dictates charge transport, and in optimizing materials for energy storage, where it influences ion conductivity and battery lifespan. Therefore, the ability to accurately predict and manipulate polymorphism represents a powerful tool for materials scientists seeking to tailor properties and unlock novel functionalities.
The swift identification of stable crystal structures is paramount in materials science, as even subtle arrangements of atoms profoundly impact a material’s functionality. OrgFlow represents a significant advancement in this field, offering a computational framework that dramatically accelerates the discovery of novel compounds with pre-defined characteristics. By accurately predicting these arrangements, researchers can bypass lengthy and costly trial-and-error synthesis, focusing instead on materials exhibiting desired properties for applications ranging from pharmaceuticals – where crystal form dictates drug bioavailability – to organic electronics demanding specific charge transport characteristics, and even next-generation energy storage solutions requiring optimized ion conductivity. This predictive capability not only reduces development time but also unlocks the potential to design materials with performance levels previously unattainable, fostering innovation across multiple scientific disciplines.
The potential for materials discovery extends significantly with a framework capable of navigating extensive chemical spaces. This computational approach doesn’t merely predict structures; it actively searches for molecules with properties ideally suited for critical technological applications. In drug development, the rapid screening of virtual compounds accelerates the identification of potential therapeutic candidates, reducing both time and cost. Simultaneously, the design of novel organic electronic materials benefits from the ability to tailor molecular arrangements for optimal charge transport and device performance. Perhaps most crucially, this expanded search capability aids in the creation of advanced energy storage solutions, such as improved battery electrolytes and electrode materials, by pinpointing compounds with enhanced stability and conductivity – ultimately paving the way for more efficient and sustainable technologies.
The efficiency of OrgFlow lies in its remarkably swift convergence to accurate crystal structure predictions; the framework consistently achieves performance nearing its peak potential with only 20 ordinary differential equation (ODE) steps. This rapid calculation speed represents a significant advancement in computational materials science, drastically reducing the time and resources required to explore complex chemical spaces. Unlike many traditional methods demanding hundreds or even thousands of iterative steps, OrgFlow’s ability to reach near-peak performance with so few steps highlights its streamlined approach and optimized algorithms, paving the way for high-throughput materials discovery and accelerating the design of novel compounds for diverse applications.
Validating computationally predicted crystal structures requires rigorous quantitative assessment, and Root Mean Square Deviation (RMSD) serves as a crucial metric for this purpose. RMSD calculates the average distance between the atoms in a predicted structure and those in a known, experimentally determined structure – essentially measuring the structural difference. A lower RMSD value indicates a higher degree of similarity and, therefore, greater confidence in the prediction’s accuracy. Beyond simple comparison, RMSD allows researchers to systematically evaluate and refine prediction algorithms, identify potential errors, and ultimately build more reliable frameworks for materials discovery. This quantitative approach is essential not only for verifying the plausibility of novel structures but also for guiding further computational and experimental investigations, ensuring that resources are focused on the most promising candidates.
The pursuit of predicting organic crystal structures, as detailed in this work with OrgFlow, reveals a fundamental truth about complex systems. The framework doesn’t impose order through rigid control, but rather allows it to emerge from the interplay of local rules governing molecular interactions. This mirrors the sentiment expressed by Henry David Thoreau: “It’s not enough to be busy; you must look to see that you’re busy with the right things.” OrgFlow, through its bond-aware loss and equivariant neural networks, focuses on the ‘right things’ – the intrinsic relationships within the molecular graph – allowing the resulting crystal structure to unfold naturally, rather than being forced into a predetermined form. The effect of the whole is not always evident from the parts; sometimes it’s better to observe than intervene, and OrgFlow embodies this principle.
What Lies Ahead?
The demonstrated capacity to generate organic crystal structures from molecular graphs represents a shift, though not necessarily a conquest. Existing methods, often calibrated for the rigid geometries of inorganic compounds, struggle with the conformational flexibility inherent to organic molecules. OrgFlow addresses this, but it does so by learning to approximate a complex, high-dimensional manifold. The true challenge isn’t simply predicting structures, but understanding why certain arrangements emerge from a sea of possibilities. Small decisions by many atoms, influenced by subtle intermolecular forces, produce global effects; control is always an attempt to override that natural order.
Future work will likely focus on expanding the scope of molecular diversity handled by these generative models. Currently, the framework excels within a defined chemical space. Extending its reach will necessitate incorporating more nuanced representations of intermolecular interactions – those fleeting, transient bonds that dictate packing arrangements. A fruitful avenue lies in exploring how these models might be coupled with experimental data, not as a means of validation, but as a source of inductive bias, guiding the generative process toward physically plausible outcomes.
Ultimately, the goal isn’t to build a perfect crystal structure predictor, but to develop a framework for understanding the principles governing self-assembly. The observed success is less about algorithmic ingenuity, and more about recognizing that order doesn’t need architects; it emerges from local rules. The limitations of any predictive model will always reflect the inherent unpredictability of complex systems.
Original article: https://arxiv.org/pdf/2602.20195.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- The Division Resurgence Best Weapon Guide: Tier List, Gear Breakdown, and Farming Guide
- GearPaw Defenders redeem codes and how to use them (April 2026)
- Last Furry: Survival redeem codes and how to use them (April 2026)
- Gold Rate Forecast
- Limbus Company 2026 Roadmap Revealed
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
- Genshin Impact Version 6.5 Leaks: List of Upcoming banners, Maps, Endgame updates and more
- The Division Resurgence Specializations Guide: Best Specialization for Beginners
- XO, Kitty season 3 soundtrack: The songs you may recognise from the Netflix show
2026-02-26 04:47