Author: Denis Avetisyan
A new framework combines the power of computational pathology with clinical data to accelerate the identification and validation of meaningful biomarkers.
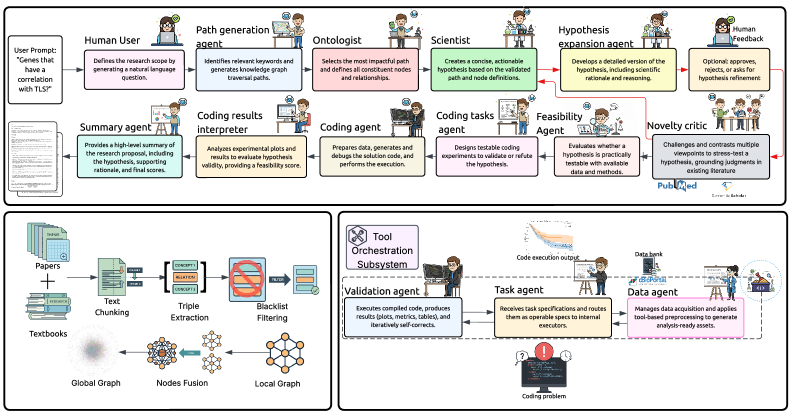
SAGE, an agentic system leveraging knowledge graphs, bridges the gap between computational pathology features, molecular biomarkers, and clinical outcomes.
Despite advances in computational pathology, a persistent challenge remains in translating AI-driven discoveries into clinically actionable insights due to a lack of transparency and biological grounding. To address this, we present ‘SAGE: Agentic Framework for Interpretable and Clinically Translatable Computational Pathology Biomarker Discovery’, a novel agentic system that correlates image-derived features with molecular biomarkers and clinical outcomes. SAGE prioritizes interpretable, biologically supported biomarkers by integrating literature-anchored reasoning with multimodal data analysis, effectively bridging the gap between novelty and clinical utility. Will this structured approach to biomarker discovery accelerate the adoption of AI in pathology and ultimately improve patient care?
The Pathology of Uncertainty
Bladder cancer presents a significant challenge to biomarker discovery due to the intricate nature of its tumor microenvironment and the limitations of conventional analytical methods. Unlike cancers with more homogeneous tissues, bladder tumors exhibit substantial variability in cellular composition, stromal elements, and immune cell infiltration, creating a complex landscape that obscures the identification of reliable biomarkers. Traditional approaches, often relying on analyzing small tissue samples or single protein markers, struggle to capture this heterogeneity and frequently yield inconsistent or misleading results. Furthermore, the throughput of these methods is often insufficient to analyze the large number of samples needed for robust validation, hindering the translation of promising biomarker candidates into clinical practice. This necessitates the development of innovative technologies capable of comprehensively profiling the tumor microenvironment with high sensitivity and throughput, ultimately paving the way for more accurate diagnostics and personalized therapies.
Analyzing whole slide images in pathology presents a significant analytical hurdle due to the natural variability within tissue samples – a phenomenon known as tissue heterogeneity. This intrinsic complexity demands advanced computational methods to discern genuine signals from background noise and artifact. Traditional manual analysis is prone to subjective interpretation, introducing variability between observers and limiting reproducibility. Consequently, researchers are increasingly employing machine learning and artificial intelligence techniques, such as deep learning convolutional neural networks, to automatically quantify morphological features and identify subtle patterns indicative of disease. These methods not only accelerate the analytical process but also offer the potential for more objective and precise diagnoses, ultimately improving patient outcomes by enabling a more nuanced understanding of disease progression and treatment response.
A significant obstacle in deciphering the intricacies of disease lies in the fragmented nature of current analytical methods; investigations frequently address genomic, proteomic, or imaging data in isolation. This compartmentalization prevents a holistic view of pathology, as these data types represent interconnected layers of biological information-genes dictate protein production, proteins influence cellular morphology observable in images, and imaging reveals the spatial organization of these molecular events. The inability to synthesize these diverse signals limits the capacity to identify truly informative biomarkers and construct predictive models, hindering progress towards personalized diagnostics and effective therapeutic strategies. A unified analytical framework, capable of seamlessly integrating these modalities, is therefore crucial to move beyond correlative studies and establish a mechanistic understanding of disease progression.
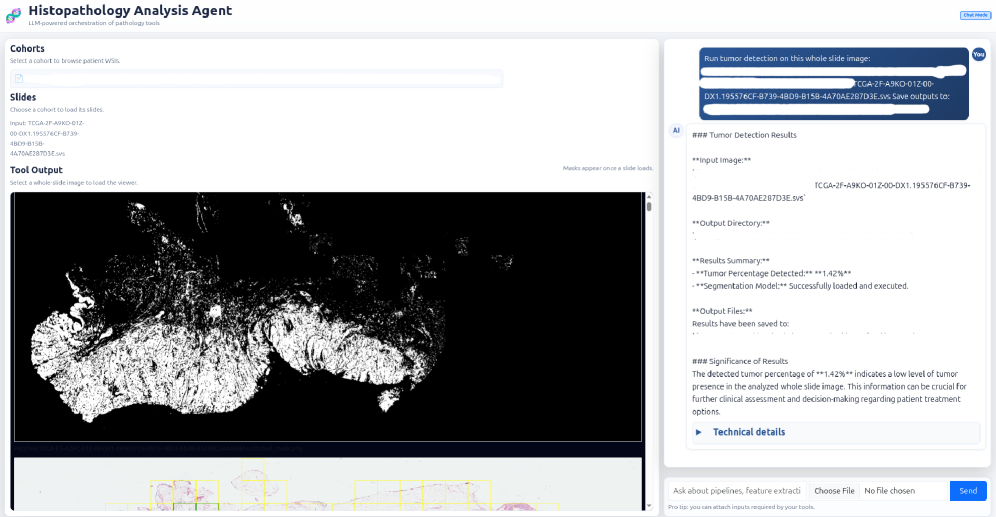
Orchestrating Discovery with Agentic Systems
SAGE utilizes a multi-agent system architecture to decompose complex biomedical discovery tasks into manageable components performed by specialized agents. This approach involves coordinating agents responsible for distinct functions, including literature review, knowledge graph construction, and hypothesis generation. Each agent operates autonomously, contributing to the overall workflow, and communicates with other agents to share information and coordinate actions. This distributed design contrasts with monolithic approaches and enables parallel processing, improved scalability, and modularity, allowing for easier updates and refinement of individual components without disrupting the entire system.
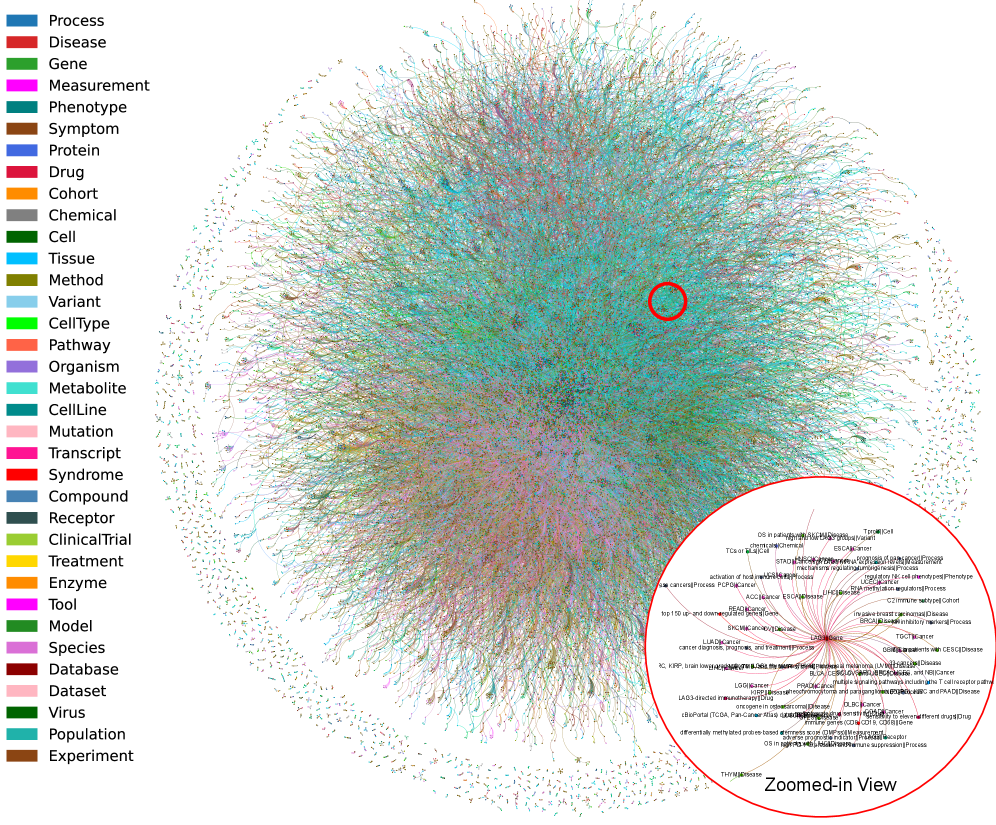
SAGE utilizes a biomedical knowledge graph constructed through automated relationship extraction from scientific literature using the GPT-4o-mini language model. This process identifies and formalizes connections between biomedical entities, such as genes, proteins, diseases, and drugs. The extracted relationships are then integrated with a pre-existing ontology, providing a structured framework for organizing and interpreting the information. This ontological foundation ensures consistency and facilitates reasoning by defining entity types, properties, and valid relationships, ultimately creating a machine-readable representation of biomedical knowledge.
SAGE utilizes large language models (LLMs) to generate hypotheses based on the constructed biomedical knowledge graph. The system refines the LLM’s reasoning process through the application of negative prompting, a technique that guides the model away from undesirable or irrelevant outputs. This approach to hypothesis generation, combined with the knowledge graph’s structured data, results in a 65% reduction in token usage compared to traditional shared-memory pipeline architectures. This improved efficiency in context management lowers computational costs and enables more scalable automated discovery processes.
Validating Signals: From Hypothesis to Evidence
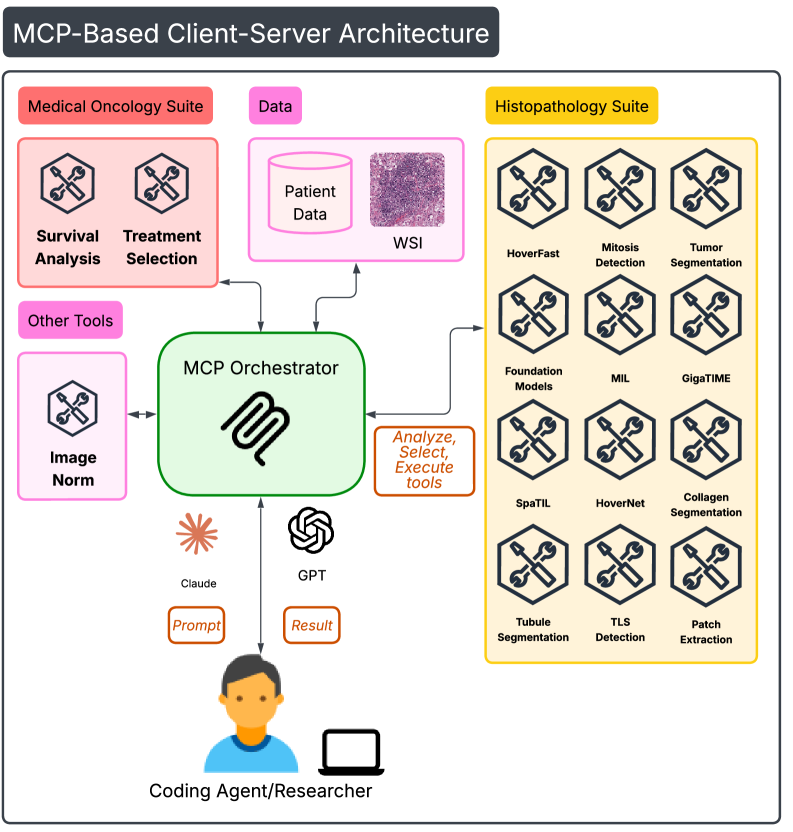
SAGE’s automated hypothesis validation utilizes a tool orchestration subsystem to streamline biomarker assessment. This system integrates established statistical methods, including survival analysis and the Cox Proportional Hazards Model, to correlate potential biomarkers with patient outcomes. The automated pipeline processes data to generate hazard ratios and p-values, enabling quantitative evaluation of biomarker significance. By automating these analyses, SAGE reduces manual effort and accelerates the identification of statistically relevant biomarkers for further investigation, providing a consistent and reproducible framework for hypothesis testing.
Rigorous testing within SAGE leverages the TCGA-BLCA (The Cancer Genome Atlas – Bladder Cancer) dataset, a publicly available resource containing genomic and clinical data from a large cohort of bladder cancer patients. This dataset is supplemented by the utilization of whole slide images (WSIs), providing detailed histopathological information for each case. The combination of genomic and imaging data allows for correlation of identified biomarkers – such as E-FABP and TLS – with clinically relevant outcomes including patient survival and disease progression. This approach ensures that biomarker candidates are not solely identified based on genomic alterations, but are demonstrably linked to observable and measurable clinical phenotypes, strengthening the translational potential of the research.
SAGE’s automated validation pipeline, applied to the TCGA-BLCA dataset, identified E-FABP and TLS as potential biomarkers for bladder cancer, demonstrating the system’s capacity for biomarker prioritization. Evaluation of SAGE’s novelty assessment revealed a high degree of concordance with expert evaluations, evidenced by a Pearson correlation coefficient of 0.91 and a Spearman rank correlation of 0.89. Quantitative error metrics further confirmed this alignment, with a Mean Absolute Error (MAE) of 0.31 and an overall error rate of 8.7% when compared to expert scores across the evaluation set.
![Patients with bladder cancer exhibiting high [latex]FABP5[/latex] expression coupled with limited tumor-infiltrating lymphocytes (TILs) demonstrate significantly reduced overall survival compared to those with low [latex]FABP5[/latex] expression and abundant TILs.](https://arxiv.org/html/2602.00953v1/x3.png)
The System’s Promise: Beyond Validation, Toward Expansion
Rigorous clinical validation, exemplified by analyses using The Cancer Genome Atlas – Bladder Cancer (TCGA-BLCA) dataset, is paramount to realizing the potential of biomarker discoveries for personalized medicine in bladder cancer. This process moves beyond simple identification, demanding confirmation that proposed biomarkers reliably correlate with disease progression, treatment response, and ultimately, patient outcomes. Utilizing TCGA-BLCA, researchers can assess biomarker performance against a large, well-characterized cohort, accounting for genomic heterogeneity and clinical diversity. Successful validation not only strengthens the biological rationale for these biomarkers but also paves the way for their integration into diagnostic assays and therapeutic strategies, enabling clinicians to tailor treatment plans to individual patient profiles and improve overall efficacy.
The conventional process of biomarker discovery is often hampered by lengthy timelines and substantial financial investment, frequently requiring years of manual data analysis and validation. However, SAGE presents a transformative solution through its fully automated pipeline, significantly diminishing both the time and cost traditionally associated with identifying clinically relevant biomarkers. By automating key steps – from data acquisition and preprocessing to statistical analysis and biomarker ranking – SAGE dramatically accelerates the transition of research findings into tangible clinical applications. This streamlined approach not only enables faster identification of potential diagnostic and therapeutic targets but also facilitates broader validation studies, ultimately paving the way for personalized medicine strategies in bladder cancer and beyond.
The strength of the SAGE platform lies not just in its success with bladder cancer, but in its adaptable design, positioning it as a broadly applicable tool for automated biomedical research. Its modular architecture permits straightforward reconfiguration for investigating diverse disease areas; researchers can readily swap datasets, adjust parameters governing genomic analysis, and refocus hypotheses without requiring extensive reprogramming. This flexibility dramatically reduces the barriers to entry for exploring novel biomarkers in conditions ranging from neurological disorders to autoimmune diseases, and even infectious disease pathology. Ultimately, SAGE promises to accelerate the pace of discovery by enabling rapid, hypothesis-driven investigations across a wide spectrum of biomedical domains, fostering a future where automated analysis complements and enhances traditional research approaches.
The pursuit of biomarker discovery, as detailed in the SAGE framework, often feels less like construction and more like tending a garden. It’s a slow revealing of interconnectedness, a coaxing of patterns from the complex interplay of computational pathology, molecular data, and clinical outcomes. This resonates with a sentiment expressed by John McCarthy: “In fact, as far as I’m concerned, the computer is the most important invention of the twentieth century.” SAGE doesn’t simply find biomarkers; it cultivates a system where hypotheses emerge from the relationships within the knowledge graph, acknowledging that scalability is often just a justification for complexity. The true value lies not in a perfect, pre-defined architecture, but in the system’s ability to adapt and reveal insights over time, even if those insights necessitate a reevaluation of initial assumptions.
The Path Forward
The architecture presented – a structured agentic system for biomarker discovery – does not solve the problem of translating computational pathology into clinical benefit. It merely relocates the inevitable points of failure. Every knowledge graph, however meticulously curated, is a map of what is believed to be true, and belief, as history repeatedly demonstrates, is a fragile foundation. The system will undoubtedly surface correlations, some statistically compelling, but discerning signal from noise remains a task not of computation, but of repeated, and often disappointing, validation.
The true challenge lies not in generating hypotheses – this system appears capable – but in gracefully accepting their falsification. The pursuit of clinical translation is, at its core, a series of carefully constructed hopes, each destined to encounter the realities of biological complexity. The system’s success will not be measured by the biomarkers it confirms, but by its capacity to rapidly and efficiently discard those that do not withstand scrutiny.
Future iterations will inevitably focus on scaling – larger datasets, more agents, increasingly sophisticated reasoning engines. But the core limitation remains unchanged: a system can only reflect the biases and gaps in the data it consumes. The most fruitful path forward may lie not in building more complex systems, but in cultivating a more humble appreciation for the limits of what any system – including this one – can truly know.
Original article: https://arxiv.org/pdf/2602.00953.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Limbus Company 2026 Roadmap Revealed
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- Total Football free codes and how to redeem them (March 2026)
- XO, Kitty season 3 soundtrack: The songs you may recognise from the Netflix show
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- The Division Resurgence Specializations Guide: Best Specialization for Beginners
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
- Guild of Monster Girls redeem codes and how to use them (April 2026)
- Gold Rate Forecast
- ‘Project Hail Mary’s Soundtrack: Every Song & When It Plays
2026-02-03 21:23