Author: Denis Avetisyan
This review explores the evolving landscape of artificial intelligence in regulatory genomics, charting a path toward models that not only predict gene function but also reveal the underlying mechanisms.
Achieving robust generalization and mechanistic insight in genomic AI requires integrating perturbation data, continual learning, and causal refinement of seq2func models.
Despite advances in machine learning, accurately predicting gene regulatory effects from DNA sequence remains a significant challenge, often failing to translate predictive power into mechanistic understanding. This review, ‘Toward Interpretable and Generalizable AI in Regulatory Genomics’, synthesizes the current state of sequence-to-function (seq2func) modeling, highlighting limitations in generalization and interpretability across diverse genetic and cellular contexts. We argue that progress hinges on reframing these models as continually refined systems, integrating targeted perturbation experiments and iterative updates through AI-experiment feedback loops. Can such self-improving systems unlock a more robust and causally grounded understanding of the cis-regulatory code, ultimately accelerating biological discovery?
Decoding the Genome: From Sequence to Cellular Behavior
Functional genomics hinges on deciphering the intricate relationship between an organism’s DNA sequence and the resulting regulatory activity within cells. The genome isn’t simply a blueprint for proteins; it’s a complex instruction manual where specific DNA sequences dictate when, where, and to what extent genes are expressed. These regulatory elements – enhancers, promoters, silencers, and insulators – bind proteins that control the transcriptional machinery, ultimately shaping an organism’s phenotype. A comprehensive understanding of how these sequences orchestrate gene expression is therefore fundamental, as it allows researchers to move beyond simply identifying genes to understanding how genetic information is actually utilized to build and maintain life. Unlocking this ‘regulatory code’ is a key step toward predicting the consequences of genetic variation and ultimately, understanding the complexities of biological systems.
Historically, dissecting the link between genomic sequence and regulatory function has proven remarkably difficult due to the inherent complexity of biological systems. Early approaches often relied on linear models – assuming a straightforward, proportional relationship between DNA features and gene activity – but these consistently failed to capture the nuanced reality. Regulatory elements rarely act in isolation; instead, they engage in intricate interactions, exhibiting synergistic or antagonistic effects. Moreover, the influence of a given sequence can be dramatically altered by its genomic context, chromatin state, and the combinatorial effects of multiple transcription factors. This non-linearity, where small changes in DNA sequence can trigger disproportionately large shifts in gene expression, has long stymied predictive efforts, necessitating the development of sophisticated computational models and large-scale experimental techniques to unravel the regulatory code.
The ability to accurately predict regulatory activity from genomic information is paramount to deciphering the complexities of gene expression and, ultimately, cellular behavior. Regulatory elements – the stretches of DNA that control when and where genes are turned on or off – orchestrate development, respond to environmental cues, and maintain cellular homeostasis. Misinterpreting these regulatory signals leads to an incomplete understanding of how genomes function, hindering progress in fields ranging from developmental biology to disease modeling. Consequently, researchers are increasingly focused on developing computational models and experimental techniques that can reliably forecast regulatory outcomes, allowing for a more complete and nuanced view of the relationship between genotype and phenotype. This predictive capability isn’t simply about identifying where regulation occurs, but also how – the strength, dynamics, and interplay of multiple regulatory factors – to truly unravel the intricate logic governing life.
![An iterative causal refinement framework leverages a pre-trained sequence-to-function model and active learning-guided perturbation assays, such as MPRA and CRISPRi, to continually refine understanding of [latex]cis[/latex]-regulatory mechanisms and improve genome-wide regulatory knowledge.](https://arxiv.org/html/2602.01230v1/x5.png)
Predicting Function: The Rise of Sequence-to-Function Models
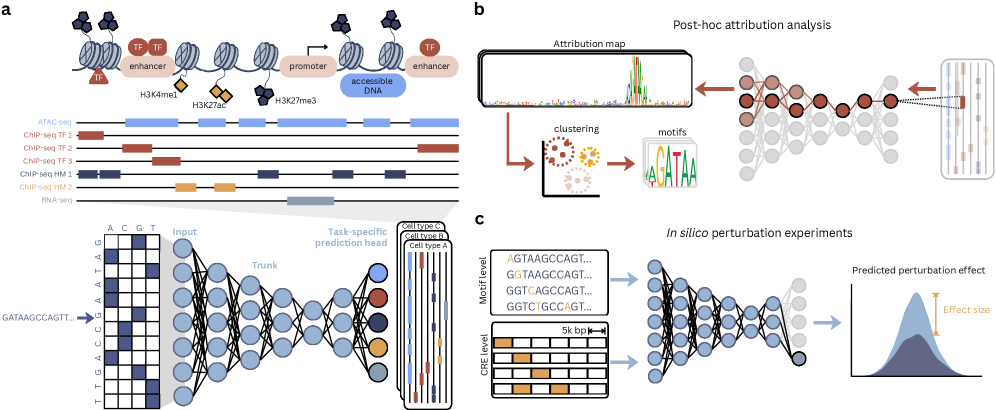
Seq2Func models represent a class of deep learning approaches designed to predict regulatory activity – such as transcription factor binding or chromatin accessibility – directly from the DNA sequence itself. Unlike traditional methods relying on handcrafted features or prior biological knowledge, these models learn predictive patterns directly from the data. Input is typically represented as a one-hot encoded sequence, where each nucleotide (A, T, C, G) is represented by a distinct binary vector. The model then processes this sequence through multiple layers of artificial neural networks to output a prediction of regulatory activity at each genomic position. This direct prediction capability allows Seq2Func models to identify novel regulatory elements and understand the complex relationship between genotype and phenotype.
Convolutional Neural Networks (CNNs) are frequently used in Seq2Func models due to their ability to identify local sequence motifs predictive of regulatory activity. Recurrent Neural Networks (RNNs), particularly LSTMs and GRUs, process DNA sequences sequentially, capturing long-range dependencies potentially important for distal regulatory element interactions. More recently, Transformer architectures, initially developed for natural language processing, have been adapted for genomic sequences, leveraging self-attention mechanisms to model relationships between distant nucleotides without the sequential constraints of RNNs; these models demonstrate parallelization advantages for training and inference on large datasets.
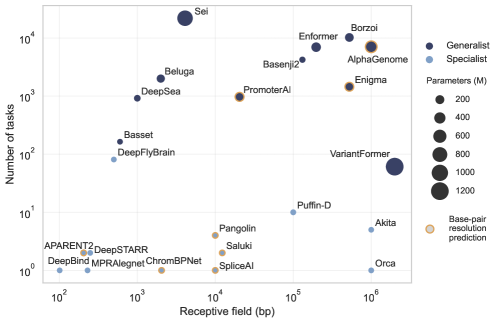
Training Seq2Func models necessitates large-scale, genome-wide profiling data, typically generated through assays like ChIP-seq, ATAC-seq, or RNA-seq, to establish relationships between DNA sequence and regulatory activity. Model performance is strongly correlated with the quantity of training data utilized; however, increasing data volume also drives the need for larger model architectures. As illustrated in Figure 2, there has been a trend toward scaling model size, measured by parameter count, from several million to billions of parameters. This scaling aims to capture complex sequence-function relationships but also introduces computational demands for both training and inference, requiring substantial computational resources and optimization strategies.
The receptive field within a Seq2Func model dictates the length of DNA sequence used as input for predicting regulatory activity. This span is critical, as regulatory elements often function through interactions across considerable genomic distances; a limited receptive field may fail to capture these distal relationships, reducing predictive accuracy. Conversely, an excessively large receptive field increases computational cost and may introduce noise from irrelevant genomic regions. The size of the receptive field is directly determined by the architecture of the model; for example, convolutional layers have receptive fields defined by kernel size, while recurrent networks accumulate information over sequential inputs, effectively expanding the considered sequence length. Optimizing the receptive field size is therefore a key consideration in model design and training to balance computational efficiency with accurate prediction of regulatory effects.
Refining Predictions: Robustness and Iterative Improvement
Model performance is susceptible to several forms of distributional shift. Data imbalance occurs when certain classes or outcomes are underrepresented in the training data, leading to biased predictions. Covariate shift arises when the input feature distribution differs between training and deployment, while label shift involves a change in the output distribution. Critically, concept shift represents a change in the relationship between inputs and outputs – the underlying concept the model is trying to learn – and is often the most challenging to address. Each of these shifts can result in decreased accuracy, reduced generalization ability, and unreliable predictions in real-world applications, necessitating ongoing monitoring and model adaptation strategies.
Active learning methodologies improve model retraining efficiency by strategically selecting data points for labeling based on their potential to reduce model uncertainty. Rather than random sampling, these techniques prioritize instances where the model exhibits low confidence or high disagreement amongst ensemble members. This targeted approach requires an iterative process: the model is initially trained on a small labeled dataset, then used to predict labels on a larger unlabeled dataset. Data points with the highest uncertainty – often determined by metrics like prediction entropy or margin sampling – are then selected for manual labeling by experts. The newly labeled data is incorporated into the training set, and the process repeats, focusing subsequent experiments on the most informative data and minimizing the labeling effort required to achieve performance gains. This contrasts with passive learning, where data is labeled randomly, and generally results in faster convergence and improved model accuracy with fewer labeled examples.
Perturbation assays, used to assess system behavior under modified conditions, commonly generate high-dimensional data due to the measurement of numerous responses at a limited set of genetic or environmental loci. This results in datasets with thousands of measurements, but relatively few variables defining the perturbation. Consequently, experimental design becomes critical to maximize information gain and minimize the cost and complexity of data analysis. Efficient designs prioritize perturbations and measurement points that provide the most significant signal regarding the system’s underlying relationships, enabling accurate model training and refinement with fewer experimental resources.
Causal refinement is an iterative process designed to enhance model accuracy by systematically identifying and addressing deficiencies through experimentation. This methodology involves initial model training, followed by the design of targeted experiments – often perturbations – to test specific causal hypotheses about the relationships between variables. Data generated from these experiments is then used to refine the model, typically by adjusting parameters or retraining with the new data. This cycle of experimentation and model updating is repeated, with each iteration focusing on areas where the model exhibits the greatest uncertainty or error, ultimately leading to improved predictive performance and a more robust understanding of the underlying system. The process relies on feedback loops to ensure continuous improvement and focuses on establishing causal relationships, rather than simply correlations.
Continual learning, also known as lifelong learning, addresses the challenge of catastrophic forgetting in machine learning models. Traditional models, when trained on a new dataset, often experience a significant decline in performance on previously learned tasks. Continual learning techniques mitigate this by enabling the model to incrementally incorporate new information without overwriting existing knowledge. Approaches include regularization-based methods that constrain weight changes, replay-based methods that store and revisit previous data, and dynamic architecture techniques that expand the model’s capacity as new tasks are encountered. The goal is to maintain a consistent level of performance across all learned tasks, effectively simulating human learning where new information is integrated without erasing prior knowledge.
![Continual learning strategies mitigate catastrophic forgetting by either encouraging convergence towards shared parameter optima [latex]⟨[/latex] or by preserving past knowledge through replay of previous examples or regularization of important parameters.](https://arxiv.org/html/2602.01230v1/x4.png)
Toward Virtual Cells and Predictive Genomics
Virtual cells represent a significant leap in computational biology, functioning as in silico models capable of simulating the intricate behavior of living cells. These models utilize Seq2Func approaches, which learn the relationship between a cell’s genetic code – its sequence – and its resulting function. By training on vast datasets of genomic information, Seq2Func models can predict how alterations to a cell’s DNA, such as mutations or gene edits, will impact its overall behavior. This predictive capability extends to forecasting changes in protein production, metabolic pathways, and even a cell’s response to external stimuli, offering researchers a powerful tool to dissect complex biological systems and test hypotheses without the need for costly and time-consuming wet-lab experiments. The ability to virtually manipulate a cell’s genome and observe the consequences holds immense promise for accelerating drug discovery and tailoring treatments to an individual’s unique genetic makeup.
Genomic language models, or GLMs, are proving essential in deciphering the intricate code of life by analyzing the statistical patterns inherent in DNA sequences. These models, akin to those used in natural language processing, learn the probability of specific DNA “words” appearing in relation to others, thereby capturing the complex relationships between genes and regulatory elements. Unlike traditional methods that focus on individual genes, GLMs consider the genome as a cohesive text, predicting how changes in one region might affect distant parts of the genome. This predictive power significantly enhances the capabilities of Seq2Func models, which simulate cellular behavior; by providing a robust understanding of the underlying genomic context, GLMs allow for more accurate predictions of gene expression and regulatory activity, ultimately pushing the boundaries of predictive genomics and personalized medicine.
The convergence of sequence-to-function models and genomic language models establishes a remarkably powerful framework for dissecting the intricacies of biological systems. By uniting the predictive capabilities of cellular simulations with the statistical insights gleaned from genomic data, researchers gain an unprecedented ability to model complex interactions within cells. This holistic methodology moves beyond analyzing individual genes or proteins, instead enabling the exploration of emergent behaviors arising from the interplay of numerous cellular components. Consequently, it facilitates the investigation of phenomena previously intractable to conventional reductionist approaches, offering new avenues for understanding development, disease, and the fundamental principles governing life itself.
The convergence of sequence-to-function models and genomic language models heralds a new era in predictive genomics, promising increasingly accurate forecasts of gene expression and regulatory activity. By computationally simulating cellular responses to genetic variations, researchers can move beyond correlation to establish more definitive links between genotype and phenotype. This capability is poised to revolutionize personalized medicine, enabling clinicians to anticipate individual patient responses to therapies and tailor treatments accordingly. Ultimately, the precise prediction of biological outcomes at the individual level will facilitate proactive healthcare strategies, potentially preventing disease onset or optimizing therapeutic efficacy – moving the focus from reactive treatment to preventative, personalized care.
The pursuit of genomic AI demands ruthless simplification. Abstractions age, principles don’t. This review champions seq2func modeling not as an end, but as a means to uncover the cis-regulatory code. Every complexity needs an alibi; thus, the integration of perturbation assays and continual learning isn’t merely about improving prediction. As Marcus Aurelius observed, “You have power over your mind – not outside events. Realize this, and you will find strength.” The article advocates for systems that refine causal understanding, mirroring this inner strength – a focus on what is essential, discarding extraneous noise to achieve robust generalization. The goal isn’t a perfect model, but a perfectly understood one.
What’s Next?
The pursuit of mechanistic understanding in regulatory genomics has, perhaps predictably, led to increasingly complex models. Yet, the enduring challenge remains not the creation of intricacy, but the distillation of essential principles. Seq2func modeling, and the related approaches detailed herein, offer a promising path, but only if tempered by a commitment to parsimony. The field must resist the allure of ever-larger datasets and more convoluted architectures, focusing instead on methods that prioritize generalization from limited, yet rigorously curated, perturbation data.
A critical, and largely unresolved, limitation lies in the static nature of current models. The genome is not a fixed entity, but a dynamic system constantly responding to internal and external cues. Future progress demands a shift towards continual learning frameworks-systems capable of refining their understanding in light of new evidence, and gracefully adapting to the inherent noise and variability of biological data. Such systems necessitate robust causal refinement strategies, moving beyond mere correlation to establish genuine mechanistic links.
Ultimately, the success of genomic AI will not be measured by predictive accuracy alone, but by its capacity to reveal the underlying logic of the cis-regulatory code. The goal is not to build black boxes that mimic biological function, but to construct transparent, interpretable models that illuminate the fundamental principles governing gene regulation. The path forward requires a willingness to embrace simplicity, and a recognition that true insight often emerges not from what is added, but from what is thoughtfully removed.
Original article: https://arxiv.org/pdf/2602.01230.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- Total Football free codes and how to redeem them (March 2026)
- Limbus Company 2026 Roadmap Revealed
- The Division Resurgence Specializations Guide: Best Specialization for Beginners
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- Brawl Stars Sands of Time Brawl Pass brings Sandstalker Lily and Sultan Cordelius sets, along with chromas and more
- Brawl Stars Brawl Cup Pro Pass arrives with the Dragon Crow skin and Chroma, unique cosmetics, and more rewards
- Clash of Clans April 2026 Gold Pass Season introduces a Archer Queen skin
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
- XO, Kitty season 3 soundtrack: The songs you may recognise from the Netflix show
2026-02-04 00:41