Author: Denis Avetisyan
A new computational pipeline combining machine learning and molecular simulations rapidly identifies promising polymer electrolytes with enhanced ionic conductivity.
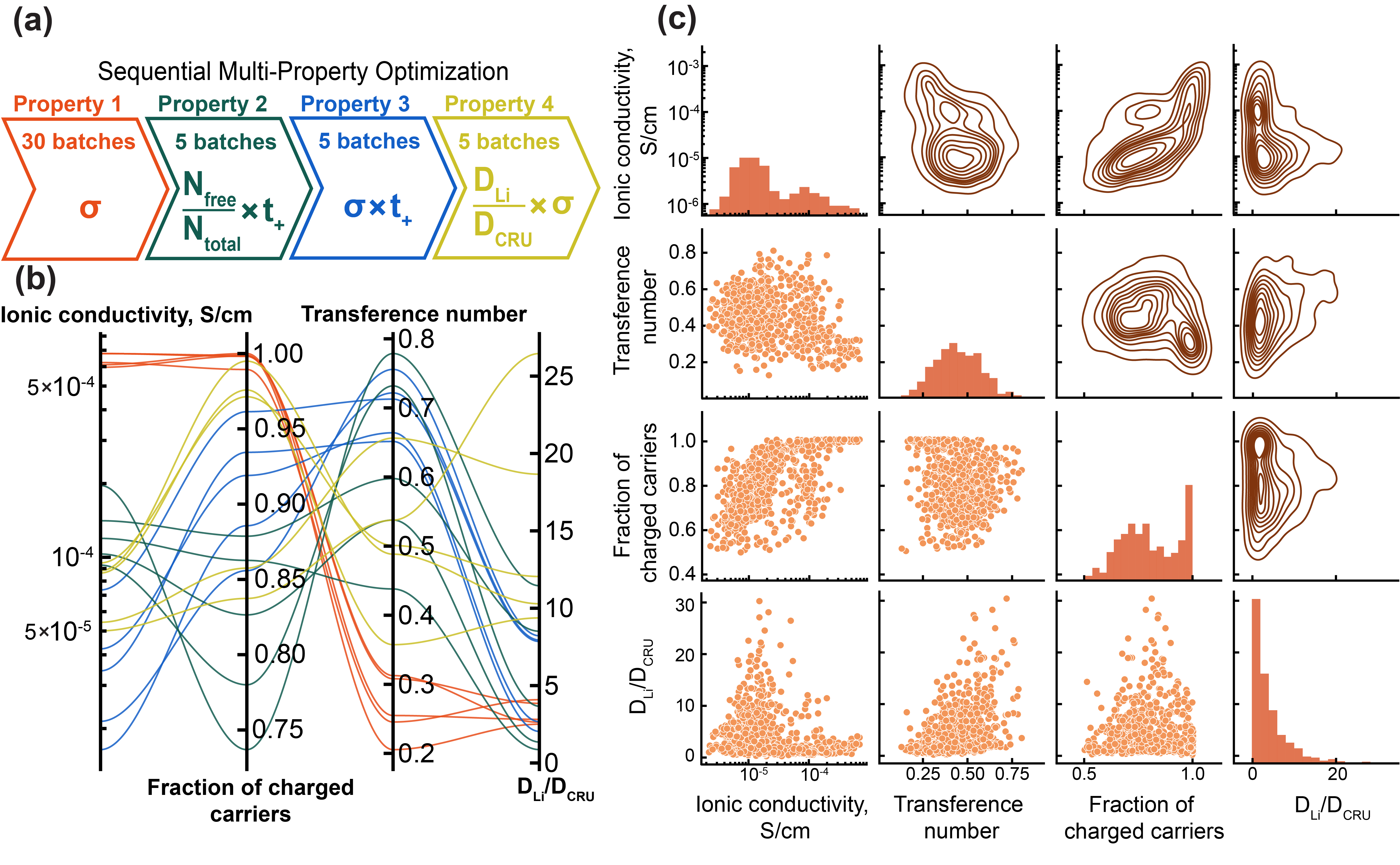
Bayesian optimization and high-throughput molecular dynamics simulations reveal branched polymers containing ketone groups as high-performing alternatives to conventional polyethylene oxide.
Achieving both high ionic conductivity and transference numbers remains a key challenge in developing safe, high-energy-density solid-state batteries. This is addressed in ‘Discovery of Polymer Electrolytes with Bayesian Optimization and High-Throughput Molecular Dynamics simulations’, which presents a high-throughput screening platform leveraging Bayesian optimization and molecular dynamics to navigate a vast chemical space of potential polymer electrolytes. The research identifies several novel candidates-particularly branched polymers incorporating ketone functional groups-that exhibit transport properties exceeding those of conventional polyethylene oxide (PEO)/LiTFSI systems. Will this computational framework accelerate the design of next-generation electrolytes for a wider range of battery technologies and multi-cation systems?
The Inevitable Decay of Electrolytes, and the Search for Graceful Transition
The widespread adoption of lithium-ion batteries is currently hampered by inherent limitations within their liquid electrolytes. These solutions, while effective at conducting ions, are flammable and prone to leakage, presenting significant safety concerns and necessitating bulky safety mechanisms. Furthermore, the electrochemical window of typical liquid electrolytes restricts the operating voltage and, consequently, the energy density achievable by the battery. This has spurred intensive research into solid-state electrolytes, materials offering the potential for enhanced safety – through non-flammability and reduced leakage – and higher energy densities by enabling the use of high-voltage cathode materials. A successful transition to solid electrolytes promises not only safer devices but also a substantial increase in the performance capabilities of batteries powering everything from portable electronics to electric vehicles.
The pursuit of next-generation, all-solid-state batteries faces a significant hurdle in the slow pace of discovering novel polymer electrolytes with sufficiently high ionic conductivity. Traditional materials science relies heavily on synthesizing and characterizing candidate materials, a process that is both time-consuming and resource-intensive. The vastness of the chemical space for possible polymer combinations presents an almost insurmountable challenge; even with high-throughput experimentation, the rate of discovery remains limited by the sheer number of potential compositions and structural arrangements. This experimental bottleneck necessitates the development of more efficient approaches, such as predictive modeling and computational screening, to accelerate the identification of promising solid electrolytes and ultimately unlock the full potential of solid-state battery technology.
The pursuit of novel solid electrolytes hinges on the ability to efficiently explore the immense landscape of potential polymer compositions, a challenge computational materials discovery aims to address. Traditional experimental methods are hampered by the sheer number of possible polymer combinations and the time-consuming nature of synthesis and characterization. Consequently, researchers are developing sophisticated algorithms and machine learning techniques to predict ionic conductivity from a material’s chemical structure, effectively narrowing the search space. These computational approaches leverage principles of materials science – such as the relationship between polymer chain flexibility, ethylene oxide content, and ion transport – to identify promising candidates before costly laboratory work begins. While accurately modeling ion dynamics within polymers remains complex, advancements in high-throughput computation and data-driven modeling are accelerating the identification of solid electrolytes with enhanced performance and safety characteristics, potentially unlocking the next generation of battery technology.
Navigating Complexity: Bayesian Optimization as a Predictive Lens
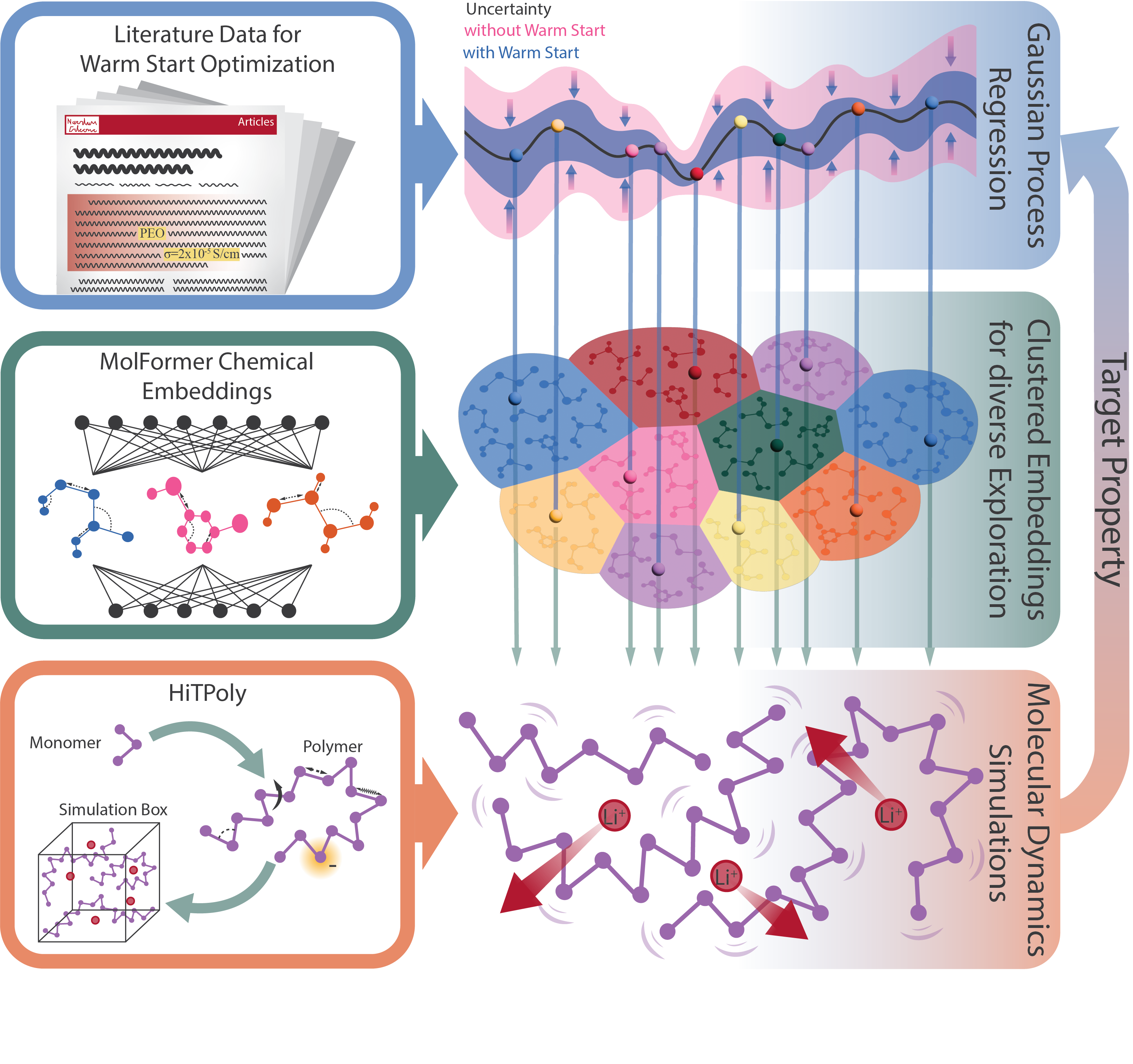
Bayesian Optimization addresses the challenge of efficiently searching the vast chemical space of polymers for materials with optimal ionic conductivity. Traditional methods often involve exhaustive screening or rely on computationally expensive simulations. Bayesian Optimization instead constructs a surrogate model – a probabilistic representation of the relationship between polymer composition and ionic conductivity – to predict performance without evaluating every possible material. This approach iteratively refines the surrogate model based on evaluation of a limited number of polymer candidates, balancing exploration of new compositions with exploitation of promising regions within the chemical space, thereby accelerating the discovery process.
The core of the optimization process relies on a Gaussian Process Regression (GPR) model to predict ionic conductivity based on a defined set of polymer features. GPR is a probabilistic, non-parametric approach suitable for modeling complex, non-linear relationships between polymer composition and resulting conductivity. The model functions by estimating a probability distribution over possible functions that fit the observed data, enabling prediction of conductivity for unseen polymer formulations. This prediction includes not only a mean value but also a measure of uncertainty, which is crucial for balancing exploration of novel chemical space with exploitation of known high-performing candidates. The GPR model is iteratively refined with each new experimental result, progressively improving its predictive accuracy and guiding the search towards polymers with optimized ionic conductivity.
MolFormer Embeddings are utilized to translate complex polymer structures into numerical descriptors suitable for input into the Gaussian Process (GP) model. These embeddings, generated via a transformer-based architecture pre-trained on a large corpus of molecular data, capture nuanced chemical information including atomic composition, connectivity, and spatial arrangements. Unlike manually engineered features, MolFormer embeddings automatically learn relevant representations directly from the polymer’s graph structure, enabling the GP model to accurately predict ionic conductivity based on these learned descriptors. The resulting high-dimensional vector representations effectively encode complex chemical relationships within the polymer, improving the predictive power of the surrogate model and accelerating materials discovery.
Principal Component Analysis (PCA) is employed to reduce the dimensionality of the feature set derived from MolFormer embeddings prior to Gaussian Process Regression. This technique identifies the principal components – orthogonal linear combinations of the original features – that capture the maximum variance in the data. By retaining only these components, while discarding those with minimal variance, the number of input features is significantly reduced. This dimensionality reduction mitigates the curse of dimensionality, improving the predictive accuracy of the GP model and reducing computational cost associated with both model training and prediction. Specifically, fewer features lead to a simpler model, reducing the risk of overfitting and accelerating the Bayesian Optimization process.
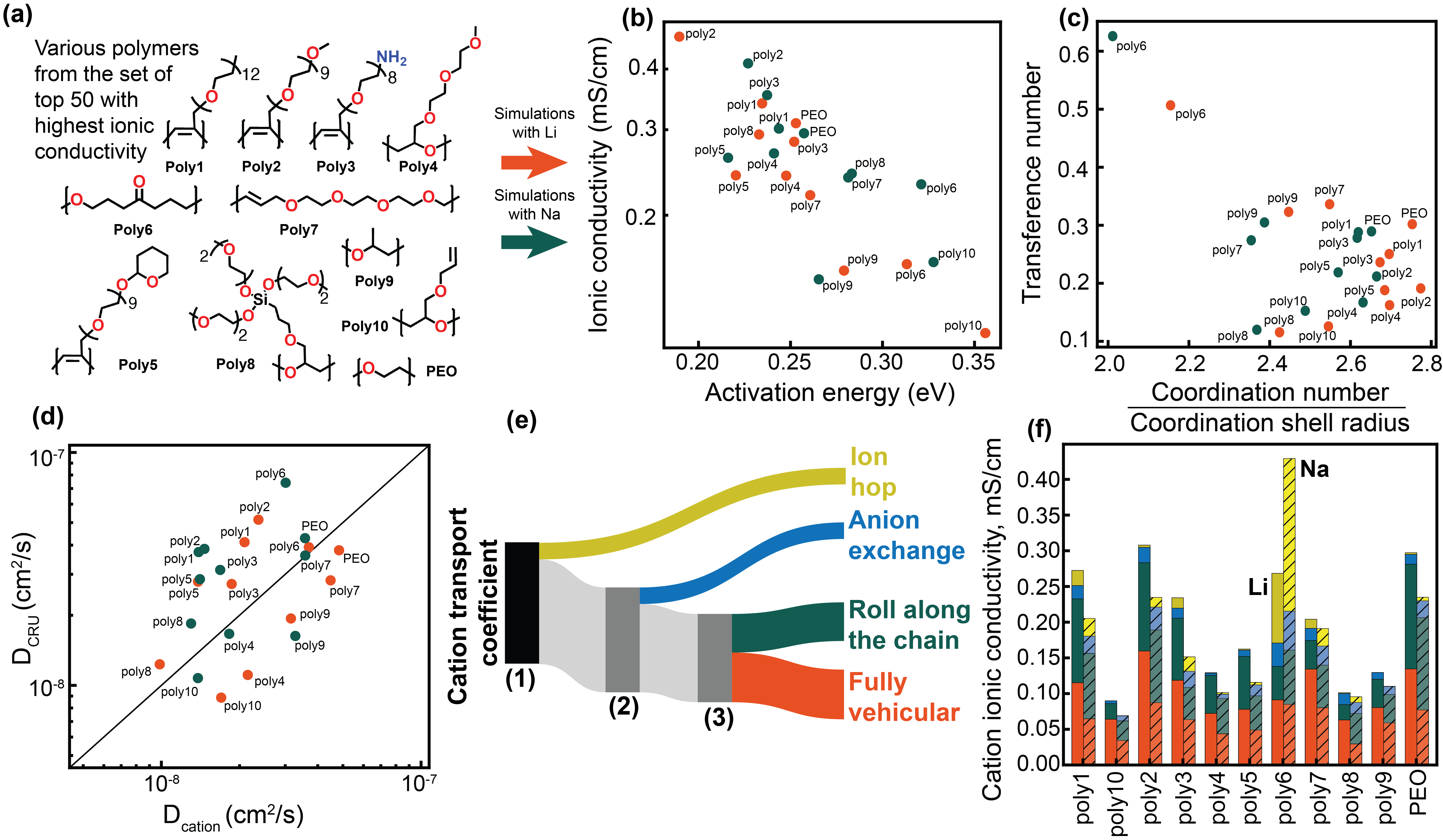
Revealing the Mechanisms: Molecular Dynamics and the Dance of Ions
Molecular Dynamics (MD) simulations utilize Newton’s equations of motion to model the time-dependent behavior of atoms and molecules, providing detailed insight into ion transport phenomena. By explicitly representing the interactions between ions and the surrounding polymer matrix, MD allows for the observation of ion hopping events – the movement of ions between coordination sites. These simulations capture not only the trajectory of individual ions but also the concurrent dynamic response of the polymer chains, including segmental motions, chain stretching, and conformational changes. Analysis of MD trajectories yields quantitative data on diffusion coefficients, ionic conductivity, and the energy landscape governing ion migration, all at the atomic scale. The technique resolves the relationship between microscopic structural features and macroscopic transport properties, complementing experimental techniques and providing a level of detail unattainable through other means.
Molecular dynamics simulations demonstrate a correlation between polymer structural features and ionic conductivity. Specifically, increased linear branching within the polymer structure leads to a demonstrable increase in ion mobility. This effect is attributed to the creation of free volume within the polymer matrix, facilitating ion hopping between segmental motions. The simulations quantify this relationship, showing that higher degrees of branching disrupt polymer chain packing, lowering the energy barrier for ion transport and ultimately enhancing the material’s ionic conductivity. Analysis of simulation trajectories reveals a statistically significant increase in ion diffusion coefficients with increasing branching density, confirming the positive influence of structural modifications on ion transport properties.
Molecular dynamics simulations demonstrate that the solvation environment significantly influences ion transport properties; ions are not isolated entities but are surrounded by solvent molecules which impact their mobility and effective size. The number and arrangement of these solvent molecules, forming a solvation shell, directly correlates with the ion’s diffusion coefficient. Furthermore, observed clustering of ions, driven by electrostatic interactions, reduces the number of available charge carriers and increases local concentration polarization, both of which decrease overall ionic conductivity. This clustering is particularly pronounced at higher ion concentrations and can lead to the formation of ion pairs or larger aggregates, effectively hindering their movement through the polymer matrix.
The HiTPoly pipeline utilizes Bayesian optimization to efficiently explore the vast compositional space of polymers for targeted ion conductivity. This involves iteratively proposing polymer formulations based on a probabilistic model-the Bayesian optimizer-and subsequently validating these predictions with molecular dynamics (MD) simulations. MD simulations quantify the ionic conductivity of the proposed polymer, providing feedback to refine the Bayesian model. This creates a closed-loop, self-correcting cycle where prediction accuracy increases with each iteration, significantly reducing the computational cost associated with materials discovery and optimization compared to traditional trial-and-error methods or exhaustive simulations.

Towards a Sustainable Future: The Promise of Enhanced Battery Technology
A new computational framework has successfully pinpointed several promising polymer electrolytes that surpass the ionic conductivity of traditional polyethylene oxide (PEO), a common but limited material in lithium-ion batteries. This breakthrough stems from a predictive model capable of efficiently screening a vast chemical space, identifying polymer structures specifically designed to facilitate faster lithium-ion transport. These newly discovered candidates demonstrate enhanced performance characteristics, potentially addressing key limitations in current battery technology and opening doors to improvements in energy density, safety, and overall efficiency. The identified polymers represent a significant step towards next-generation batteries capable of powering electric vehicles with extended range and enabling more reliable grid-scale energy storage solutions.
Optimization of novel polymer electrolytes benefits significantly from informed modeling strategies, as demonstrated by a recent study employing Gaussian Process (GP) regression. The research team implemented a ‘Warm Start’ approach, initializing the GP model with existing experimental data rather than relying on a purely random starting point. This technique allowed the model to rapidly converge on promising electrolyte candidates, substantially outperforming a standard GP model lacking this pre-existing knowledge. The ‘Warm Start’ effectively guides the optimization process, focusing computational resources on regions of chemical space likely to yield high ionic conductivity, thereby accelerating materials discovery and reducing the need for extensive trial-and-error experimentation. This method represents a powerful advance in the rational design of next-generation battery materials.
Analysis of promising polymer electrolytes revealed a striking correlation between molecular structure and ionic conductivity. Researchers observed a significant enrichment of linear branches within the chemical structures, with these groups appearing 57 times more frequently than other chemical features. This oversampling strongly suggests that the presence of these linear segments plays a crucial role in facilitating ion transport within the material. The findings indicate that prioritizing linear branching during the design of new polymer electrolytes could be a highly effective strategy for enhancing battery performance and achieving higher energy densities, representing a pivotal step towards next-generation energy storage solutions.
The pursuit of enhanced battery technology stands to redefine energy storage, and recent advancements directly address limitations within current lithium-ion systems. Improved polymer electrolytes, identified through a novel optimization framework, promise batteries that are not only more energy-dense – enabling extended ranges for electric vehicles and greater efficiency in portable devices – but also considerably safer. Traditional lithium-ion batteries utilize flammable liquid electrolytes, posing risks of thermal runaway and fire; solid polymer electrolytes mitigate these dangers. This increased safety, coupled with the potential for higher energy density, is critical for widespread adoption of electric vehicles and for the reliable storage of renewable energy on a grid scale, facilitating a transition towards a more sustainable energy future. These developments represent a significant step towards batteries capable of meeting the growing demands of a rapidly electrifying world.
The research presented exemplifies a systematic approach to material discovery, mirroring the inevitable entropy of existing solutions. Just as systems age, so too do conventional polymer electrolytes reach performance limits. This study doesn’t simply iterate on known structures, but actively seeks alternatives through a computationally driven pipeline. As Jean-Paul Sartre noted, “Existence precedes essence.” In this context, the process of high-throughput screening and Bayesian optimization-the ‘existence’-defines the emergent properties-the ‘essence’-of the novel polymer electrolytes. The identification of branched polymers with ketone groups demonstrates how a deliberate method can yield materials exceeding the capabilities of established standards, offering a potential solution to the decay inherent in current technology.
The Inevitable Refinement
The presented work, a successful marriage of computational efficiency and materials discovery, merely identifies promising candidates – it does not, and cannot, conjure permanence. The identified branched polymers with ketone groups will, like all things, degrade. The question is not if their conductivity will diminish, but when, and at what rate. This is not a failing of the research, but a fundamental property of existence. The pipeline itself, however, represents a more enduring achievement-a method for systematically exploring a vast chemical space, acknowledging that stability is often just a temporary reprieve from entropy.
Future iterations will undoubtedly refine the machine learning models, incorporating more nuanced descriptors of polymer behavior. Yet, the true challenge lies not in achieving ever-greater predictive accuracy, but in understanding the limits of prediction itself. Can a model, however sophisticated, truly account for the unpredictable interplay of microscopic defects, environmental stressors, and the sheer passage of time? The pursuit of “ideal” electrolytes risks becoming a Sisyphean task, ignoring the inevitability of material fatigue.
A more fruitful avenue may lie in embracing impermanence. Could future research focus on designing electrolytes that gracefully degrade, perhaps releasing benign byproducts or undergoing predictable structural changes? Such an approach acknowledges the transient nature of materials, shifting the focus from absolute stability to controlled evolution. The aim is not to halt decay, but to manage it-to accept that all systems age, and to prepare for the inevitable.
Original article: https://arxiv.org/pdf/2602.17595.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-22 19:51