Author: Denis Avetisyan
A new framework efficiently assesses the stability of complex nanoparticles, minimizing the need for extensive computational simulations.
![The analysis of an [latex]Al_{70}Co_{10}Fe_5Ni_{10}Cu_5[/latex] decagonal quasicrystalline alloy reveals a layer-dependent structure, where atomic composition, electronegativity, valence electron concentration, and coordination number vary systematically across six topologically defined layers, indicating a gradient in chemical and electronic properties throughout the nanoparticle.](https://arxiv.org/html/2602.17528v1/x1.png)
This review details a machine learning approach leveraging topological layer embeddings and gradient-boosted decision trees to predict nanoparticle configurational stability using data from density functional theory calculations.
Predicting the stability of nanoparticles remains a significant challenge due to the vast configurational space arising from complex atomic environments. This is addressed in ‘Interpretable Machine Learning of Nanoparticle Stability through Topological Layer Embeddings’, which introduces a data-efficient machine-learning framework leveraging a novel, layer-resolved descriptor to decompose nanoparticles into surface, intermediate, and core regions. By coupling this descriptor with gradient-boosted decision trees and a ranking-based learning strategy, the research demonstrates accurate stability prediction with limited density functional theory calculations. Can this physically interpretable framework accelerate materials discovery and guide the exploration of complex nanoparticle configurations through active learning strategies?
The Challenge of Alloy Stability: A Systems Perspective
The development of novel metallic alloys, such as the Al70Co10Fe5Ni10Cu5 system, hinges on accurately predicting their thermodynamic stability – a process vital for tailoring material properties. However, conventional computational approaches, often relying on exhaustive searches of potential configurations, prove remarkably demanding in terms of processing time and resources. This computational expense arises because predicting stability requires considering a multitude of atomic arrangements and their associated energies, effectively creating a high-dimensional search space. Consequently, the pace of materials discovery is often limited by the prohibitive cost of accurately assessing alloy stability using these traditional methods, motivating the pursuit of more efficient predictive techniques.
Alloy stability isn’t simply a matter of elemental composition; rather, it’s profoundly shaped by the arrangement of atoms within the material. Chemical disorder – the deviation from perfectly ordered atomic arrangements – introduces energetic fluctuations that can either stabilize or destabilize a given alloy phase. Simultaneously, surface segregation, where certain elements preferentially accumulate at the alloy’s exterior, alters the overall free energy and impacts thermodynamic behavior. Adding to this complexity is coordination topology, which describes how atoms connect to their neighbors – different bonding arrangements affect stability through variations in electronic structure and strain. These interconnected factors create a high-dimensional energy landscape where predicting the most stable alloy configuration requires navigating a vast array of possibilities, making accurate prediction a considerable scientific challenge.
The sheer complexity of metallic alloy configurations presents a formidable challenge to stability prediction. Each atom within an alloy possesses numerous potential arrangements, influenced by its interactions with neighboring elements and the overall structural framework. This results in a vast, multi-dimensional configuration space where even modestly sized alloys contain an astronomical number of possible atomic arrangements – far exceeding the capacity of any computational method to perform an exhaustive search. Consequently, researchers must rely on approximations and intelligent algorithms to navigate this space efficiently, focusing on the most probable and energetically favorable configurations while acknowledging the inherent limitations in fully capturing the alloy’s true thermodynamic state. The scale of this computational burden underscores the need for innovative approaches that can effectively prune the configuration space and accelerate the identification of stable alloy structures.
![Principal component analysis of Al70Co10Fe5Ni10Cu5 decagonal quasicrystalline alloy nanoparticles reveals a non-linear relationship between descriptor variance and energetic stability, and demonstrates that an Optuna-optimized XGBoost model outperforms baseline regression models in predicting nanoparticle ranking based on [latex]E_{tot}[/latex].](https://arxiv.org/html/2602.17528v1/x2.png)
Reframing Stability: A Problem of Relative Ranking
Traditional approaches to nanoparticle stability prediction often involve calculating the absolute energy of each configuration, a computationally expensive process susceptible to systematic errors. Instead, this work reframes the problem as one of relative ranking; the goal is to correctly order nanoparticle configurations based on their inherent stability without predicting precise energy values. This allows the model to focus on discerning the relative differences in stability between configurations, a task more amenable to machine learning algorithms designed for ranking problems. By evaluating the model’s ability to correctly order configurations – determining which is more stable than another – we bypass the need for accurate absolute energy prediction and can leverage algorithms optimized for ordinal relationships.
Formulating nanoparticle stability as a ranking problem enables the application of machine learning models specifically designed for ordering tasks, circumventing the limitations of traditional regression approaches. Regression models predict continuous values – absolute energies in this case – which can be sensitive to systematic errors and require precise calibration. Conversely, ranking models focus on predicting the relative order of stability between different configurations. This shift simplifies the learning task; the model only needs to determine which configuration is more stable than another, rather than predicting an exact energy value. Consequently, ranking models are generally more robust to errors in the underlying data and can achieve higher accuracy with smaller training datasets, particularly when absolute energy scales are less critical than identifying the most stable configurations.
Density Functional Theory (DFT) calculations serve as the foundational data for establishing the accuracy of the proposed ranking approach. Specifically, DFT computes the total energy of each nanoparticle configuration, providing a definitive, albeit computationally expensive, measure of its stability. These energies are then used to generate a ground truth ranking – configurations with lower DFT energies are considered more stable and placed higher in the ranking. This ground truth ranking is essential for two primary purposes: training machine learning models to predict relative stability and validating the performance of these models by comparing their predicted rankings against the established DFT-derived rankings. The accuracy of the machine learning models is therefore directly dependent on the quality and reliability of the initial DFT calculations.
![Training set size significantly improves the ranking of AlFeCoNiCu nanoparticle configurations, as evidenced by increasing Spearman rank correlation [latex]ho[/latex], screening recall, and decreasing regret-measured as the energy difference between the model’s top-[latex]k[/latex] predictions and the true global minimum-across test sets of size 10, 20, and 30, with screening budgets of [latex]k=5[/latex] and [latex]k=10[/latex].](https://arxiv.org/html/2602.17528v1/x5.png)
Layer-Resolved Descriptors: Dissecting Atomic Environments
The Layer-Resolved Descriptor functions by partitioning a nanoparticle’s atomic structure into discrete topological layers, defined by the radial distance from a designated nanoparticle center. This decomposition allows the model to differentiate between atoms located at the surface, in the core, or within intermediate layers. Consequently, the descriptor captures variations in the local atomic environment-specifically, coordination number, bond distances, and angles-as a function of radial position. This spatially-resolved information is then used as input features for machine learning, enabling the model to discern how atomic arrangements within different layers contribute to the overall stability of the nanoparticle configuration.
The layer-resolved descriptor generates numerical features representing the atomic environment of nanoparticles, which are then inputted into an XGBoost regression model to predict the relative stability of different atomic configurations. XGBoost, a gradient-boosted decision tree algorithm, iteratively builds an ensemble of decision trees, weighting each tree based on its predictive performance on the training data. This approach allows the model to capture non-linear relationships between the layer-resolved descriptor features and the potential energy of the configurations, ultimately providing a quantitative assessment of stability. The model outputs a predicted energy value for each configuration, enabling ranking and comparison of relative stabilities.
The methodology incorporates an active learning loop to optimize the efficiency of density functional theory (DFT) calculations. Instead of randomly selecting configurations for DFT evaluation, the XGBoost model predicts the uncertainty of each candidate structure; those with the highest uncertainty are then prioritized for DFT calculation. This iterative process, where the model is continuously refined with new DFT data, significantly improves predictive accuracy while minimizing the total number of computationally expensive DFT calculations required. Performance metrics demonstrate a Spearman Rank Correlation of ≥ 0.97 in predicting relative stability, achieved with training datasets as small as 200-300 DFT-calculated structures, indicating a substantial reduction in computational cost compared to traditional methods.
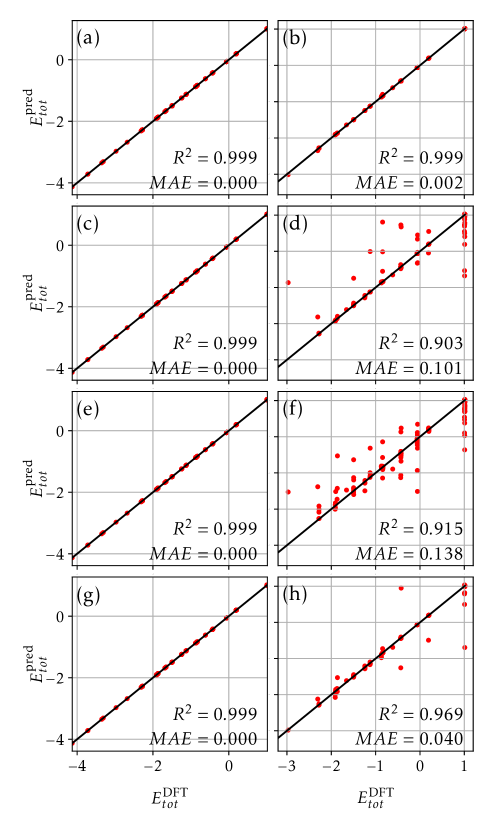
Decoding Stability: SHAP Analysis and Feature Importance
SHAP (SHapley Additive exPlanations) analysis was instrumental in deciphering the complex relationships within the XGBoost model and pinpointing the primary factors influencing predicted alloy stability. This method, rooted in game theory, assigns each feature a value representing its contribution to a specific prediction, allowing researchers to move beyond simple feature importance rankings. The analysis revealed that certain structural characteristics – notably atomic packing fractions and specific interatomic distances – consistently exerted the strongest influence on stability predictions. By quantifying these contributions, SHAP analysis not only validated the model’s internal logic but also provided valuable insights into the fundamental principles governing alloy formation, potentially guiding the discovery of novel, stable materials.
The XGBoost model’s predictive power extends beyond simply identifying stable alloys; through SHAP analysis, researchers can dissect why certain structural characteristics contribute to stability. This quantification reveals that features representing atomic packing and electronic configurations-specifically, the variance in atomic distances and the density of states near the Fermi level-are consistently the most influential determinants. The analysis demonstrates a clear relationship between specific structural motifs and stability, indicating that alloys with tightly packed structures and favorable electronic properties are inherently more likely to be stable. This insight moves beyond empirical observation, offering a data-driven understanding of alloy behavior and paving the way for the rational design of novel, stable materials with targeted properties.
Rigorous validation confirms the predictive power of the XGBoost model, demonstrating a remarkable capacity to rank alloy configurations by stability. Assessment via Spearman Rank Correlation reveals a high degree of accuracy in ordering these configurations, while Top-5 Recall approaches unity, indicating the model consistently identifies stable structures within the top five predictions. Crucially, this performance is achieved with limited training data – only 200 to 300 structures are required – and is further substantiated by near-zero regret, signifying minimal deviation from optimal predictions when applied to unseen data. This efficiency and accuracy highlight the model’s potential for accelerating materials discovery by effectively prioritizing promising alloy compositions.
Accelerated Discovery: Top-k Screening and the Future of Materials Design
The development of a trained machine learning model facilitates a process known as Top-k Screening, a powerful technique for swiftly narrowing down the vast landscape of potential nanoparticle compositions. Rather than exhaustively evaluating every conceivable alloy, this approach leverages the model’s predictive capabilities to identify the k most promising candidates for detailed examination. This prioritization is achieved by scoring each material based on desired properties, allowing researchers to concentrate resources on those with the highest likelihood of success. Consequently, Top-k Screening dramatically reduces the computational demands of materials discovery, enabling the rapid exploration of complex compositional spaces and accelerating the identification of novel materials with targeted characteristics.
Traditional materials design relies heavily on computationally expensive simulations to predict the properties of numerous alloy combinations, creating a significant bottleneck in the discovery process. This new approach circumvents this limitation by employing machine learning to pre-screen a vast chemical space, dramatically reducing the number of computationally demanding first-principles calculations required. By focusing resources on only the most promising candidates – those predicted to exhibit desirable characteristics – researchers can accelerate the identification of novel alloys with tailored properties. This targeted strategy not only saves substantial computing time and energy, but also opens doors to exploring more complex alloy compositions that were previously inaccessible due to computational constraints, ultimately fostering a faster and more efficient materials innovation cycle.
The convergence of machine learning and first-principles calculations represents a paradigm shift in materials science, enabling a more deliberate and streamlined approach to alloy development. Traditionally, discovering new materials relied heavily on trial-and-error experimentation or computationally expensive simulations. This new methodology utilizes machine learning algorithms to predict the properties of numerous potential nanoparticle compositions, effectively narrowing the search space. These predictions are then rigorously validated using first-principles calculations – quantum mechanical simulations grounded in the fundamental laws of physics – ensuring accuracy and reliability. By intelligently combining predictive power with foundational physics, researchers can efficiently identify and prioritize the most promising candidates, significantly reducing both the time and resources required to bring novel materials to fruition and accelerating innovation in diverse fields.
The pursuit of predictable system behavior, as demonstrated by this research into nanoparticle stability, echoes a fundamental tenet of design. If the system survives on duct tape-represented here by computationally expensive first-principles calculations-it’s likely overengineered. This work’s layer-resolved descriptor, enabling efficient predictions with limited data, suggests a move towards elegant simplicity. As Jean-Jacques Rousseau observed, “Good manners are born from self-respect.” Similarly, a well-designed machine learning framework respects the underlying physics, deriving predictive power not from brute force, but from a clear understanding of the whole-the interplay between layers and the resulting configurational stability-rather than focusing solely on isolated parts.
Beyond the Surface
The predictive capacity demonstrated by this framework, while promising, rests upon a foundation of descriptors-representations of complex systems distilled into numerical values. It is a familiar reduction, and one always fraught with peril. One does not repair a failing lung by charting its surface area; the intricacies of gas exchange, the vascular network, the cellular interactions-these are the governing factors. Similarly, future work must move beyond merely identifying stable configurations, and begin to elucidate why those configurations are stable, linking descriptor space to the underlying physics with greater fidelity.
The active learning strategy employed here is a crucial step, acknowledging the impracticality of exhaustively mapping the potential energy landscape. Yet, it highlights a fundamental tension: efficiency often comes at the cost of completeness. The selected training points, however intelligently chosen, inevitably introduce a bias. The system, after all, can only ‘learn’ what it is shown. Future iterations should explore methods to quantify and mitigate this bias, perhaps through adversarial training or the incorporation of known physical constraints.
Ultimately, the true test will not be the ability to predict stability, but to design stability. To move beyond correlation and towards control. This requires not just a map of the landscape, but an understanding of the forces that shape it. The challenge, then, is to build a framework that is not merely predictive, but generative-one that can propose novel nanoparticle structures with pre-defined properties.
Original article: https://arxiv.org/pdf/2602.17528.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-22 02:58