Author: Denis Avetisyan
A new approach uses artificial intelligence to predict how changing a molecule’s structure will affect its properties, accelerating the search for better drug candidates.
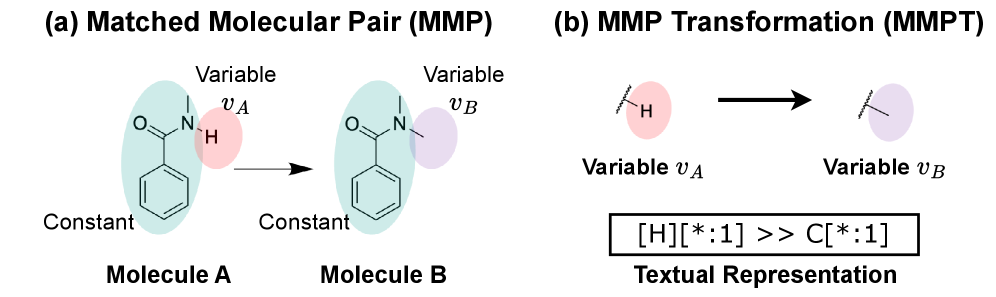
This review details a retrieval-augmented framework leveraging foundation models to perform matched molecular pair transformations for efficient analog generation in medicinal chemistry.
Designing novel molecules with desired properties remains a significant challenge, often hindered by a lack of control over specific chemical edits. This is addressed in ‘Retrieval-Augmented Foundation Models for Matched Molecular Pair Transformations to Recapitulate Medicinal Chemistry Intuition’, which introduces a framework that learns transformations between molecules based on matched molecular pairs (MMPs). By leveraging foundation models and a retrieval-augmented generation (RAG) approach, the authors demonstrate improved diversity, novelty, and controllability in generating analog structures. Could this method accelerate the discovery of promising drug candidates by more effectively mirroring medicinal chemistry intuition?
The Illusion of Molecular Innovation
Historically, the design of new molecules – a process known as analog generation – has been constrained by approaches that prioritize predictability over true innovation. These traditional methods frequently depend on established chemical rules or the laborious screening of vast compound libraries, effectively limiting exploration to well-trodden areas of chemical space. This reliance on pre-defined parameters can stifle the discovery of genuinely novel structures with potentially superior properties. While ensuring chemical feasibility, these techniques often fail to venture beyond incremental modifications of existing compounds, hindering the identification of breakthrough molecules that lie outside the scope of conventional design principles. Consequently, a significant portion of the potentially beneficial chemical universe remains unexplored due to the inherent limitations of these established analog generation strategies.
The pursuit of novel molecules hinges on the ability to systematically explore chemical space, often achieved through matched molecular pair transformations – subtle modifications to existing compounds designed to improve properties or activity. However, accurately predicting the impact of these seemingly minor changes presents a significant computational hurdle. Each transformation requires evaluating the complex interplay of atomic and electronic effects, demanding intensive calculations that quickly become prohibitive as the number of potential modifications grows. While conceptually straightforward – altering one functional group or adding a single atom – the resulting impact on a molecule’s behavior is rarely predictable from first principles, necessitating large datasets and sophisticated machine learning models to efficiently identify viable candidates from the vast landscape of possibilities. This computational cost frequently limits the scope of molecular innovation, pushing researchers to prioritize focused libraries over truly expansive exploration.
The sheer intricacy of molecular structure presents a formidable challenge to innovation in chemistry and materials science. Each molecule isn’t simply a collection of atoms, but a three-dimensional arrangement influencing its properties and interactions – a landscape where even minor alterations can yield dramatic changes. Consequently, exhaustively searching for novel compounds with desired characteristics through traditional methods proves impractical, as the number of possibilities expands exponentially with each added atom. Intelligent algorithms and computational techniques are therefore essential to efficiently traverse this vast ‘chemical space’, prioritizing promising candidates and accelerating the discovery of new materials, drugs, and technologies. These methods move beyond random screening, instead employing predictive models and machine learning to navigate complexity and pinpoint molecules with the greatest potential before costly and time-consuming physical synthesis and testing even begin.

A Foundation Built on Patterns
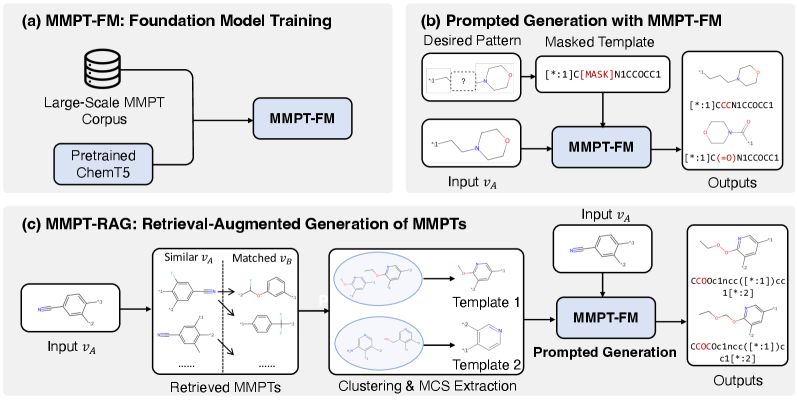
MMPT-FM is a foundation model developed through training on an extensive dataset of molecular variations, specifically focusing on examples of variable substitutions within chemical structures. This training methodology allows the model to predict probable and chemically valid modifications to existing molecules and to generate novel molecular structures based on learned patterns. The model’s capability stems from its ability to analyze and replicate the relationships between molecular components and the effects of substituting one functional group for another, effectively enabling in silico molecular design and optimization. The scale of the training data is crucial, providing MMPT-FM with a broad understanding of chemical space and facilitating the generation of realistic and synthetically accessible molecular modifications.
MMPT-FM utilizes the Transformer architecture, a neural network design known for its parallel processing capabilities and effectiveness in sequence modeling. This foundation is further strengthened by initialization from ChemT5, a pre-trained model specifically designed for chemical language understanding and generation. This transfer learning approach allows MMPT-FM to inherit ChemT5’s existing knowledge of chemical syntax, semantics, and relationships between molecular substructures. Consequently, MMPT-FM demonstrates enhanced proficiency in processing and generating valid and chemically meaningful molecular representations, effectively adapting strong language modeling capabilities to the domain of chemical structures.
During inference, MMPT-FM employs Beam Search to efficiently identify optimal molecular substitutions. This algorithm maintains a user-defined number of candidate sequences – the “beam” – at each step of the substitution process. Rather than selecting the single most probable substitution at each position, Beam Search explores multiple high-probability options in parallel. Each candidate sequence is scored based on its predicted properties and likelihood, and the algorithm iteratively expands and prunes the beam, retaining only the most promising candidates. This process allows MMPT-FM to move beyond greedy decoding and discover substitutions that maximize desired characteristics, even if those substitutions are not immediately apparent based on local probabilities.
Context is King (Even for Algorithms)
MMPT-RAG builds upon the MMPT-FM architecture by incorporating a Retrieval-Augmented Generation (RAG) framework to leverage information from external reference datasets during molecule generation. This process involves retrieving relevant molecular examples from these datasets based on similarity to the input query, and then conditioning the generative model on both the query and the retrieved examples. By grounding generation in existing, validated molecular data, MMPT-RAG aims to improve the fidelity and relevance of generated molecular analogs, effectively expanding the knowledge base accessible during the generation process beyond the model’s internal parameters.
The MMPT-RAG framework employs the Hierarchical Navigable Small World (HNSW) algorithm to construct an efficient nearest neighbor index for rapid retrieval of relevant molecular contexts. HNSW facilitates approximate nearest neighbor search by building a multi-layer graph where each layer represents a successively coarser approximation of the data. This hierarchical structure allows the algorithm to quickly navigate the graph, identifying candidate nearest neighbors with low latency. The index stores vector representations of molecular structures, enabling similarity searches based on distance metrics, and significantly accelerating the process of finding molecules similar to a given query structure compared to exhaustive search methods. This efficient retrieval is crucial for providing the generative model with relevant examples during the augmentation phase.
Molecular representations for retrieval are generated using both Morgan Fingerprints and Maximum Common Substructure (MCS) analysis. Morgan Fingerprints, also known as Extended Connectivity Fingerprints (ECFPs), provide a circular representation of an atom’s environment, capturing structural features and allowing for rapid similarity comparisons via bit vector operations. Complementarily, MCS analysis identifies the largest shared substructure between molecules, offering a more precise, though computationally intensive, method for determining structural similarity. The combination of these two approaches allows for both efficient broad-spectrum searching using fingerprints and focused identification of highly similar analogs via MCS, improving the accuracy of retrieved molecular contexts for the generation process.
MMPT-RAG enhances molecular analog generation by incorporating retrieved examples as conditional input during the generative process. This retrieval-augmented approach demonstrably improves the quality, diversity, and chemical validity of generated molecules compared to models operating without external context. Specifically, the system achieved a state-of-the-art recall of up to 46.81% when evaluated on a cross-patent generation task, indicating a significant increase in the ability to retrieve and utilize relevant prior art during analog creation. This performance metric reflects the model’s success in generating novel molecules that are both chemically plausible and structurally similar to existing compounds within the retrieved dataset.
![MMPT-RAG generates responses by clustering retrieved variables, extracting a representative message condition signal (MCS) for each cluster, and then conditioning generation on each cluster's [latex] ext{MCS}[/latex].](https://arxiv.org/html/2602.16684v1/x8.png)
The Illusion of Progress, and Where We Go From Here
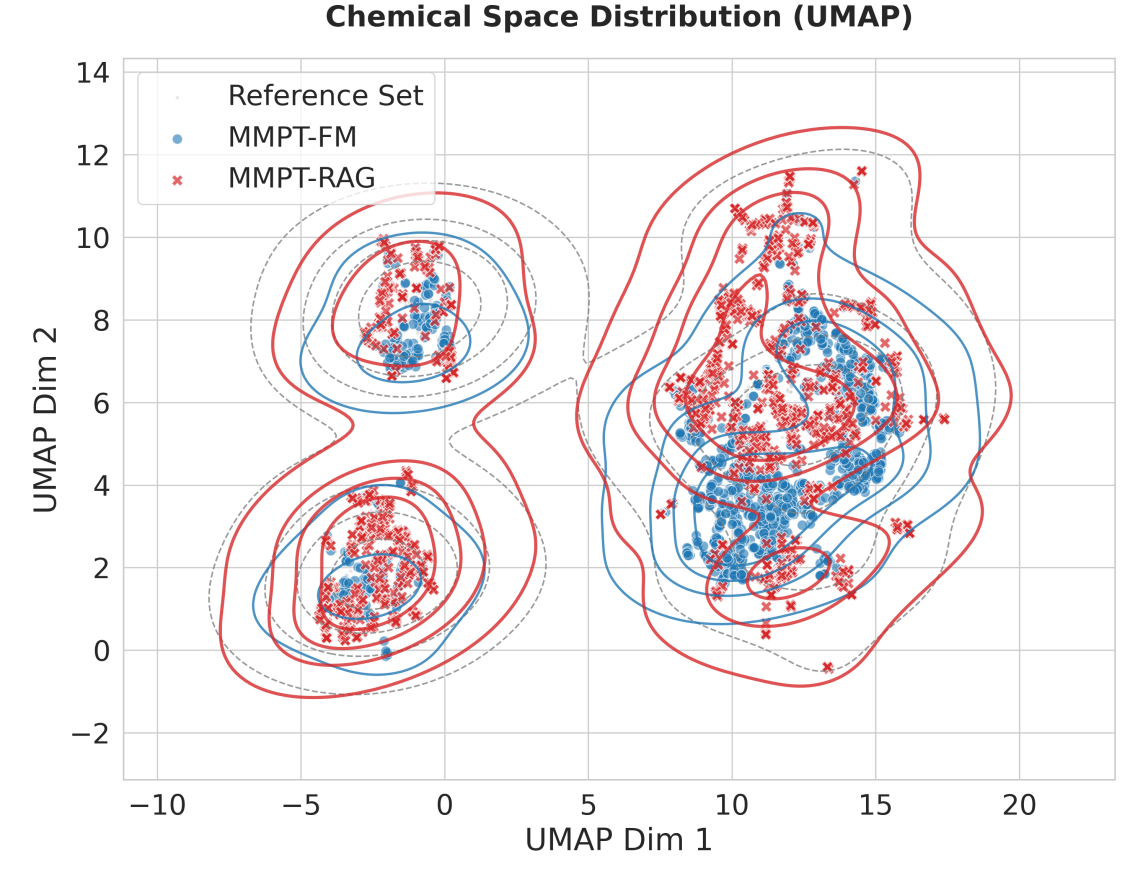
The creation of novel molecules, a cornerstone of both pharmaceutical and materials innovation, is often hampered by the time-consuming nature of generating and evaluating potential candidates. Recent advancements in machine learning have begun to address this bottleneck, and the MMPT-RAG framework demonstrates a significant leap forward in analog generation – the process of creating molecules similar to a known starting point. By rapidly producing a diverse array of structurally related compounds, MMPT-RAG dramatically accelerates the exploration of chemical space, allowing researchers to investigate a far greater number of possibilities in a given timeframe. This speed is achieved through a retrieval-augmented generation approach, enabling the model to efficiently build upon existing chemical knowledge and quickly propose viable candidates for further scrutiny, ultimately shortening the discovery cycle and potentially unlocking breakthroughs in various scientific fields.
The generation of structurally diverse yet chemically valid compounds is now significantly streamlined through this novel framework, offering substantial benefits to both drug discovery and materials science. By efficiently exploring a vast chemical space, the model accelerates the identification of potential candidates with desired properties, as demonstrated by its 82.1% recall on established, in-distribution tasks. This high recall indicates a robust ability to retrieve relevant compounds, reducing the need for extensive and costly physical synthesis and testing. The framework’s capacity to propose varied molecular structures – while adhering to chemical plausibility – not only speeds up the initial stages of research but also broadens the scope of potential innovations in fields reliant on molecular design.
Further development of the framework centers on integrating increasingly sophisticated parameters crucial for real-world applications. Current research aims to move beyond purely structural generation and incorporate predictive elements like target binding affinity – assessing how strongly a generated molecule interacts with a specific biological target – and ADMET properties, which encompass absorption, distribution, metabolism, excretion, and toxicity. By directly optimizing for these critical factors during the generative process, the model promises to yield not only novel chemical structures, but also compounds with a higher probability of success as drug candidates or functional materials, thereby significantly streamlining the discovery pipeline and reducing costly late-stage failures.
Continued development of the MMPT-RAG framework hinges on strategically augmenting its foundational data and refining the mechanisms by which relevant information is retrieved. Current evaluations demonstrate a Recall-o – the ability to generate valid compounds outside of the training set – of 12.99%, alongside a Novelty score of 30.1%, indicating substantial, yet improvable, potential for discovering truly unique chemical entities. Increasing the diversity and volume of training data promises to broaden the model’s understanding of chemical space, while optimizing retrieval algorithms will enable more efficient access to pertinent molecular information. These combined enhancements are projected to not only bolster the framework’s overall performance but also significantly expand its applicability to a wider range of complex challenges in areas like drug design and materials innovation.
The pursuit of elegant solutions in generative chemistry invariably runs headlong into the brick wall of production realities. This work, framing analog generation as learned molecular transformations, feels predictably optimistic. It proposes a retrieval-augmented framework to guide foundation models – a clever enough construct, though one can already anticipate the scaling nightmares of maintaining a truly comprehensive retrieval database. As Tim Berners-Lee observed, “This is not about technology; it’s about people.” The model’s success hinges not on the sophistication of the algorithm, but on the quality and consistency of the data fed into it. If a bug in the retrieval process is reproducible, then at least the system is predictably unstable, a small comfort in a field built on controlled approximations.
What’s Next?
This exercise in reframing analog generation as learned molecular transformations feels… familiar. It’s a sophisticated wrapper, admittedly, but one built atop the eternally frustrating problem of chemical representation. The framework neatly sidesteps some limitations of direct generative models, yet simply pushes the difficulty onto the retrieval stage. How robust is this ‘augmentation’ to noisy or incomplete datasets? Production chemistry will inevitably reveal edge cases – the subtly different reaction conditions, the unrecorded impurities – that no amount of foundation model training can anticipate.
The real challenge, as always, isn’t generating a molecule, but generating the right molecule, one that scales, is manufacturable, and doesn’t suddenly fail in phase II trials. This work hints at an interesting direction, but the devil, predictably, resides in the details of validation. Demonstrating genuine improvement over existing MMPT methods – beyond carefully curated benchmark datasets – will be the true test.
One suspects future iterations will focus on increasingly complex retrieval mechanisms, perhaps incorporating experimental data directly. But ultimately, it’s a safe bet that the next ‘revolutionary’ approach will introduce a fresh set of unforeseen problems. Everything new is just the old thing with worse docs.
Original article: https://arxiv.org/pdf/2602.16684.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-19 21:08