Author: Denis Avetisyan
A new review assesses the current limitations of AI-powered drug discovery systems and charts a path toward more effective, agentic frameworks.
![A significant chasm exists between the streamlined, computationally-focused workflows currently dominating early-stage drug discovery-reliant on representations like [latex]SMILES[/latex] strings, curated databases, and text mining-and the complex, multi-modal biological realities of iterative wet lab experimentation and multi-objective optimization inherent in bringing therapies to fruition, a limitation this work seeks to address through architectural innovation.](https://arxiv.org/html/2602.10163v1/x1.png)
Current agentic AI systems for drug discovery face critical gaps, particularly in peptide therapeutics and multi-objective optimization, requiring new design principles for integrating diverse computational methods.
Despite recent advances in artificial intelligence, translating the promise of agentic systems into practical drug discovery remains challenging. This is the central question addressed in ‘Beyond SMILES: Evaluating Agentic Systems for Drug Discovery’, where we benchmark six leading frameworks across diverse tasks-from peptide therapeutics to resource-constrained scenarios-revealing critical capability gaps in areas like protein language model integration and multi-objective optimization. Our analysis suggests these limitations stem from architectural choices rather than fundamental knowledge deficits, indicating a potential to unlock significantly improved performance. Can next-generation frameworks be designed to function as true computational partners, effectively navigating the complexities of modern drug development under realistic constraints?
The Rising Cost of Innovation: A Drug Discovery Paradox
Despite exponential gains in computational power and the application of increasingly sophisticated algorithms, the process of discovering new drugs is experiencing a counterintuitive rise in both cost and duration. Historically, bringing a single new pharmaceutical to market took roughly ten years and cost approximately $1.2 billion; current estimates suggest these figures have ballooned, exceeding fifteen years and nearly $2.6 billion. This paradox stems not from a lack of technological advancement, but from the escalating complexity of modern drug targets and the sheer volume of data requiring analysis. While computers can rapidly screen millions of compounds in silico, translating these digital predictions into viable clinical candidates remains a significant hurdle, demanding extensive and costly laboratory validation and clinical trials. The increased regulatory scrutiny and the need to address increasingly complex diseases further contribute to this growing time and cost burden, highlighting a critical inefficiency in the current pharmaceutical innovation pipeline.
The inherent intricacy of biological systems presents a formidable challenge to modern drug discovery, contributing to alarmingly low success rates. Unlike simpler systems, living organisms exhibit vast networks of interacting components – genes, proteins, metabolites – where perturbing one element can trigger cascading and often unpredictable effects. Furthermore, researchers now grapple with an explosion of diverse data types – genomic sequences, proteomic profiles, imaging data, electronic health records – each offering a fragmented view of the biological reality. Integrating these disparate datasets, and accurately modeling the complex interplay within biological systems, remains a major hurdle; current computational approaches frequently oversimplify these interactions, leading to promising candidates that ultimately fail in clinical trials due to unforeseen complexities and off-target effects. This struggle to reconcile data diversity with system complexity underscores a critical need for more sophisticated analytical tools and modeling techniques capable of capturing the holistic nature of life.
Modern drug discovery is increasingly hampered not by a lack of data, but by an inability to effectively synthesize it. While biological research generates vast datasets from disparate fields – encompassing genomic sequences, proteomic analyses, and clinical trial results – current computational frameworks struggle to integrate these diverse sources into a cohesive understanding. A recent assessment reveals a significant capability gap: existing systems can fully address zero out of fifteen defined knowledge integration tasks crucial for successful drug development. This ‘knowledge integration bottleneck’ prevents researchers from identifying meaningful patterns, formulating robust hypotheses, and ultimately, accelerating the discovery of novel therapeutics, despite advances in data generation technologies.

Intelligent Automation: Orchestrating Discovery with Agency
An agentic framework addresses inefficiencies in drug discovery by integrating and managing a variety of computational tools – including molecular modeling software, simulation platforms, and data analysis pipelines – into a cohesive automated system. This orchestration moves beyond simple task automation to enable a dynamic workflow where tools are intelligently sequenced and their outputs are used as inputs for subsequent processes. The framework’s ability to coordinate these diverse resources accelerates the identification of potential drug candidates, reduces the need for manual data transfer and analysis, and ultimately shortens the overall drug development timeline by automating steps from initial target validation through lead compound optimization.
WorkflowAutomation within an agentic framework for drug discovery utilizes software to define, execute, and monitor a series of interconnected tasks, thereby establishing streamlined pipelines. This automation reduces the need for manual data transfer and repetitive operations, increasing throughput and minimizing the potential for human error. Specifically, it involves the automated execution of processes such as high-throughput screening data analysis, in silico modeling, and report generation. The implementation of WorkflowAutomation results in quantifiable efficiency gains, including decreased task completion times and a reduction in operational costs associated with manual intervention, ultimately accelerating the drug development timeline.
LLMOrchestration functions as the central control mechanism within agentic automation for drug discovery, utilizing Large Language Models (LLMs) to dynamically manage and coordinate automated workflows. This involves the LLM interpreting experimental data, evaluating progress against defined objectives, and subsequently selecting and initiating appropriate computational tools – encompassing tasks such as molecular modeling, simulations, and data analysis – to progress from initial target identification through hit discovery, lead optimization, and ultimately, preclinical candidate selection. The LLM’s role extends beyond simple task execution to include adaptive workflow adjustments based on real-time results, effectively creating a closed-loop system that minimizes manual intervention and accelerates the overall discovery timeline.
![A shift from current LLM-centric architectures, which rely on a single language model to orchestrate tools via API calls, to a multi-paradigm approach employing a coordinator to manage diverse computational paradigms-including [latex] ext{ML training pipelines}[/latex], [latex] ext{RL optimization loops}[/latex], and [latex] ext{physics simulations}[/latex]-enables independent execution and aggregated decision-making.](https://arxiv.org/html/2602.10163v1/x5.png)
Bridging Disparate Data: A Holistic View of Biological Systems
Modern drug discovery relies on the convergent analysis of diverse data modalities to comprehensively characterize disease mechanisms and therapeutic targets. Genomics provides insights into genetic predispositions and disease pathways, while proteomics details protein expression and post-translational modifications, offering a functional view of cellular processes. Clinical data, encompassing patient demographics, medical history, and treatment responses, contextualizes these molecular-level findings with real-world outcomes. Integrating these data types – genomic, proteomic, and clinical – allows for the identification of biomarkers, the prediction of drug efficacy, and the stratification of patient populations, ultimately increasing the probability of successful drug development and personalized medicine.
Data Modality Integration (DMI) establishes a unified knowledge base by combining data from diverse sources, such as genomic, proteomic, and clinical datasets. Knowledge Graph Integration (KGI) is a key technique used within DMI, representing data entities and their relationships as a graph structure. This allows for complex queries and inferences across modalities that would be difficult or impossible with siloed data. The resulting knowledge base facilitates data harmonization, resolving inconsistencies and enabling a comprehensive view of biological systems and disease mechanisms. Successful DMI, leveraging KGI, requires standardized data formats and robust data governance to ensure data quality and reproducibility.
The integration of multi-modal data sources – genomics, proteomics, clinical trials, and others – significantly enhances peptide representation and, consequently, the accuracy of predictions within drug discovery. By linking peptide sequences to associated biological effects and clinical outcomes, researchers can move beyond sequence-based predictions to model more complex relationships. This holistic approach allows for improved target identification, enhanced prediction of drug efficacy and toxicity, and facilitates a more rational design of peptide therapeutics. Specifically, integrated datasets enable the development of machine learning models capable of identifying subtle patterns and correlations that would be undetectable when analyzing single data modalities, leading to more informed decision-making at each stage of the drug development process.
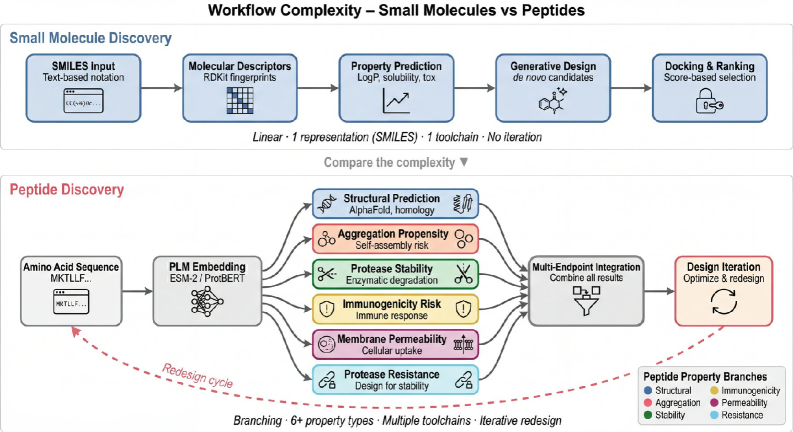
Navigating Complex Objectives: Multi-Objective Optimization and Reinforcement Learning
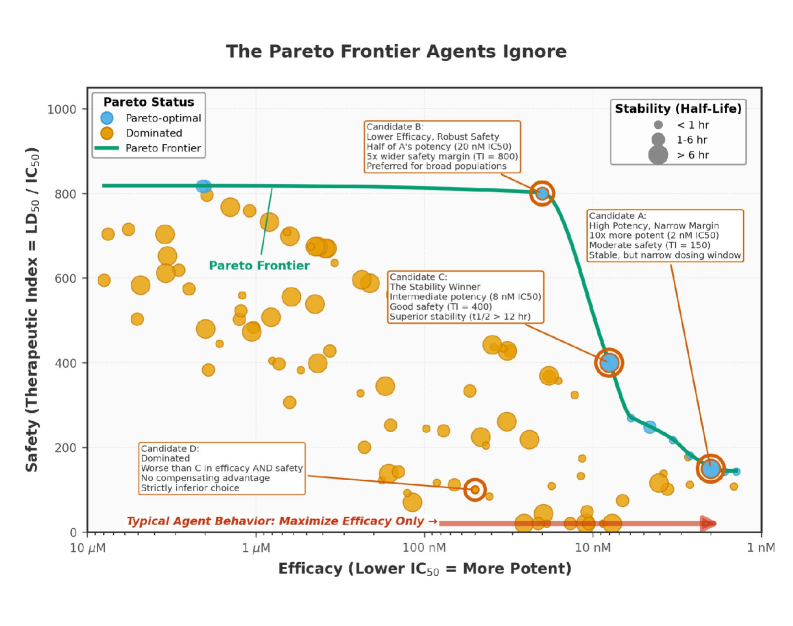
Drug discovery presents inherent challenges due to the simultaneous and often conflicting requirements of maximizing therapeutic efficacy, minimizing adverse safety profiles, and controlling manufacturing or research and development costs. These competing objectives necessitate the application of MultiObjectiveOptimization (MOO) techniques, which differ from traditional single-objective optimization by seeking a set of Pareto-optimal solutions rather than a single best solution. A Pareto-optimal solution is one where no objective can be improved without simultaneously worsening at least one other objective. MOO algorithms, such as weighted sum methods, ε-constraint methods, and evolutionary algorithms, are employed to identify this Pareto front, allowing researchers to explore the trade-space and select compounds that best balance these critical attributes based on specific priorities and risk tolerance.
Reinforcement Learning (RL) applied to drug discovery utilizes an agentic framework where an ‘agent’ learns to iteratively select actions – in this case, modifications to molecular structures – within a defined chemical space. The agent receives rewards based on predicted or experimentally determined outcomes, such as efficacy and safety, allowing it to optimize a policy for navigating the design space. This learning process enables the agent to identify sequences of actions that maximize cumulative rewards, effectively discovering compounds with improved characteristics. The optimization isn’t limited to a single objective; the RL framework can handle multiple, potentially conflicting, objectives by assigning weights or utilizing Pareto optimization techniques to identify a set of optimal solutions representing the best trade-offs.
MultiParadigmOrchestration in drug discovery integrates computational techniques to broaden the scope of molecular exploration and improve the accuracy of biological activity predictions. Specifically, GenerativeModeling, often utilizing techniques like Variational Autoencoders or Generative Adversarial Networks, creates novel chemical structures with desired properties. These in silico generated compounds are then evaluated using InVivoModeling – computational simulations that mimic biological processes – to predict their behavior within a living system. This combined approach allows researchers to efficiently assess a larger chemical space than traditional methods, identifying potential drug candidates with optimized characteristics and reducing the need for extensive laboratory synthesis and testing.
Accelerating Discovery with Limited Resources: The Power of Active Learning
When experimental resources are scarce, ActiveLearning emerges as a pivotal strategy within agentic frameworks designed to accelerate scientific progress. Rather than passively acquiring data, this technique enables an agent to intelligently select the most valuable data points for labeling – those predicted to yield the greatest increase in model accuracy. This targeted approach dramatically reduces the demand for costly and time-consuming experiments, allowing researchers to achieve significant insights with a fraction of the typical resource investment. By prioritizing information gain, ActiveLearning effectively amplifies the impact of limited resources, making impactful discovery attainable even in constrained environments and broadening access to innovation beyond well-funded laboratories.
Active learning represents a paradigm shift in experimental design, particularly crucial when facing budgetary or logistical constraints. Instead of passively acquiring data across an entire dataset, this approach intelligently prioritizes which data points require manual labeling by human experts. By focusing on the most informative samples – those likely to yield the greatest improvement in model accuracy – active learning dramatically reduces the volume of expensive and time-consuming experiments needed to achieve a desired outcome. This targeted data acquisition isn’t simply about efficiency; it allows researchers to maximize the value of each experiment, accelerating the pace of discovery and opening opportunities for innovation in resource-limited environments where traditional, exhaustive approaches are impractical.
The convergence of automated workflows and intelligent optimization strategies holds considerable promise for accelerating drug discovery, particularly in environments where resources are limited; however, current implementations face significant hurdles. While the potential exists to broaden access to innovation, present frameworks demonstrate support for a strikingly narrow range of tasks – successfully addressing only 0 of 15 defined classes. Furthermore, the agreement between expert evaluations and automated scoring remains weak, as evidenced by a Kappa score of just 0.22, indicating substantial discrepancies and highlighting the need for improved algorithms and validation processes before these systems can reliably contribute to impactful discoveries in resource-constrained settings.
![Despite demonstrating comparable reasoning abilities for both small molecules and peptides-with no statistically significant difference in mean scores (95% CI: [-0.255, 0.02])-current agentic frameworks fail to leverage this peptide knowledge through dedicated tools.](https://arxiv.org/html/2602.10163v1/x2.png)
The pursuit of agentic AI in drug discovery, as detailed in the paper, highlights a critical need to move beyond simply optimizing for speed or yield. Any algorithm prioritizing efficiency at the expense of addressing the complexities of peptide therapeutics – a notoriously difficult area – carries a societal debt. This resonates with Thomas Kuhn’s observation that, “science does not proceed by accumulating additions to existing knowledge, but by occasionally replacing old paradigms with new ones.” The current paradigm in agentic systems often treats optimization as a purely technical problem, overlooking the ethical implications of automating workflows that demand nuanced understanding and careful consideration of multi-objective challenges. Truly transformative agentic AI requires a shift – a new paradigm – that embeds ethical considerations directly into the design process.
The Road Ahead
The pursuit of agentic systems for drug discovery, as this work illuminates, has largely focused on replicating existing workflows, rather than fundamentally reimagining the process. Scaling computation without a concurrent scaling of ethical and epistemological rigor is, frankly, a crime against the future. Current frameworks excel at optimizing for single objectives-predictable, measurable outcomes-but fall short when confronted with the inherent messiness of biological systems and the need for multi-objective optimization. Every algorithm encodes a worldview, and the implicit bias towards easily quantifiable traits must be addressed.
The limitations regarding peptide therapeutics are particularly telling. The field’s preoccupation with SMILES notation-a reductionist representation of molecular structure-reveals a deeper tendency to prioritize computational convenience over biological nuance. A truly agentic system will require a shift towards more expressive representations, integrating protein language models not merely as prediction engines, but as active participants in the design process.
The next generation of these frameworks must therefore prioritize interpretability, robustness, and-most crucially-value alignment. It is no longer sufficient to simply discover potential drug candidates; the system must articulate why those candidates are promising, and demonstrate an understanding of the trade-offs involved. The challenge is not merely technical, but philosophical: can an algorithm be designed to embody the principles of responsible innovation?
Original article: https://arxiv.org/pdf/2602.10163.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Invincible Season 4 Episode 4 Release Date, Time, Where to Watch
- Physics Proved by AI: A New Era for Automated Reasoning
- Gold Rate Forecast
- American Idol vet Caleb Flynn in solitary confinement after being charged for allegedly murdering wife
- Magicmon: World redeem codes and how to use them (March 2026)
- “Wild, brilliant, emotional”: 10 best dynasty drama series to watch on BBC, ITV, Netflix and more
- Total Football free codes and how to redeem them (March 2026)
- Goddess of Victory: NIKKE 2×2 LOVE Mini Game: How to Play, Rewards, and other details
- Invincible Creator on Why More Spin-offs Haven’t Happened Yet
- Simulating Humans to Build Better Robots
2026-02-12 10:54