Author: Denis Avetisyan
A new generative AI framework harnesses the power of crystal symmetry to dramatically improve the prediction of stable and novel materials.
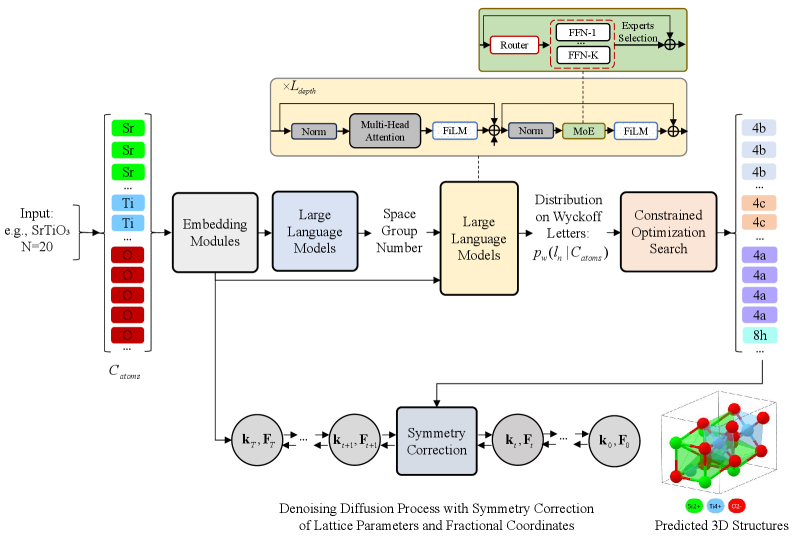
This work introduces a symmetry-driven diffusion model that enforces fine-grained geometric constraints for rigorous crystal structure prediction, leveraging Wyckoff positions and space groups.
Predicting stable crystal structures remains a significant challenge, often hindered by reliance on existing databases or approximate symmetry handling. This limitation motivates the work ‘Universal Fine-Grained Symmetry Inference and Enforcement for Rigorous Crystal Structure Prediction’, which introduces a novel framework leveraging large language models to directly infer and enforce crystallographic symmetry during the generative process. By guiding diffusion models with predicted Wyckoff patterns and rigorously maintaining algebraic consistency, this approach achieves state-of-the-art performance in materials discovery benchmarks. Could this symmetry-driven generative paradigm unlock access to entirely uncharted regions of materials space, bypassing the constraints of prior knowledge?
The Intractable Complexity of Crystalline Symmetry
The sheer combinatorial complexity inherent in crystal structure prediction presents a formidable challenge. Investigating every conceivable arrangement of atoms within a material-even for relatively simple chemical compositions-quickly becomes computationally intractable. This vast “chemical space” expands exponentially with increasing unit cell size and the number of atoms considered, meaning a brute-force approach is simply not feasible. Traditional CSP methods, therefore, grapple with balancing the need for thorough exploration against the limitations of available computing power, often requiring significant approximations or focusing on limited regions of this expansive landscape to yield results within a reasonable timeframe. This constraint hinders the discovery of truly novel and potentially valuable crystalline materials.
Crystal Structure Prediction (CSP) fundamentally relies on exploiting the symmetry inherent in crystalline materials, specifically through the application of Space Group symmetry; however, this seemingly beneficial approach presents significant computational hurdles. Precisely defining and enforcing these symmetry constraints during structure optimization is remarkably expensive, demanding substantial processing power and time. The complexity arises because algorithms must not only identify the correct symmetry operations but also ensure they are consistently applied throughout the search for the lowest energy structure. Furthermore, numerical inaccuracies and the limitations of computational methods can lead to violations of symmetry, resulting in incorrect or unrealistic structures. Consequently, researchers often face a trade-off between computational feasibility and the accuracy of symmetry implementation, frequently resorting to approximations that compromise the reliability and predictive power of CSP methods.
Many crystal structure prediction methods, while increasingly sophisticated, often compromise on fully accounting for crystallographic symmetry to manage computational demands. These approaches frequently employ simplifying assumptions or heuristic rules when applying space group constraints, effectively limiting the search to a subset of potentially viable arrangements. While expediting calculations, this simplification can inadvertently exclude genuinely stable, yet non-intuitive, crystal structures-hindering both the accuracy of predictions and the discovery of truly novel materials. The reliance on approximations, therefore, represents a fundamental trade-off between computational efficiency and the potential for groundbreaking materials discovery, prompting ongoing research into more robust and efficient symmetry-handling techniques.
![Our method surpasses DiffCSP++ in predicting stable, unique, and novel compositions of [latex]BaP_3[/latex] (with N=16), as evaluated by the SUN metrics established by Zeni et al.](https://arxiv.org/html/2602.17176v1/x3.png)
A Framework Rooted in Symmetry and Composition
The proposed framework generates crystal structures by integrating a Diffusion Model with Large Language Models (LLMs). The process is conditioned on both the chemical composition of the desired material and its predicted Space Group symmetry. The Diffusion Model, responsible for the iterative construction of the crystal structure, is guided by the compositional input and the Space Group, which dictates the symmetry constraints. This conditioning ensures that generated structures adhere to crystallographic principles and reflect the specified chemical formula, effectively linking chemical composition to three-dimensional atomic arrangements.
The framework employs a Large Language Model (LLM) to predict the Space Group associated with a given chemical composition. This prediction serves as a critical conditioning element for the subsequent generative process. The LLM is trained on a dataset of known chemical compositions and their corresponding Space Groups, enabling it to infer the most likely symmetry for a novel composition. By incorporating the predicted Space Group, the generative model is constrained to produce structures consistent with that symmetry, significantly reducing the search space and improving the validity of generated crystal structures. This approach allows the framework to move beyond purely data-driven generation and leverage known crystallographic principles.
The prediction of compatible Wyckoff letters is a critical component of ensuring the structural validity of generated crystal structures. Wyckoff letters denote the symmetry-equivalent positions within a space group, defining where atoms can reside while maintaining the structure’s symmetry. The Large Language Model (LLM) predicts these letters based on the inferred space group and chemical composition, effectively narrowing the possible atomic arrangements to those consistent with the crystal’s symmetry. This prediction constrains the subsequent diffusion model’s generation process, preventing the creation of structurally invalid or unrealistic configurations by limiting the search space to only those positions allowed by the designated Wyckoff positions.
Architectural Innovations: Scaling Capacity with Symmetry Modulation
The models utilize a Transformer architecture, a neural network design excelling at sequence modeling, and augment it with a Soft Mixture of Experts (SoftMoE) layer. SoftMoE introduces multiple expert sub-networks within each Transformer layer, with a gating network dynamically routing input tokens to a subset of these experts. This selective activation increases the model’s effective capacity-the number of parameters contributing to a given computation-without a proportional increase in computational cost. Instead of activating all parameters for every input, only the activated experts contribute, enabling the model to scale to larger sizes – up to 1.1 trillion parameters in some configurations – while maintaining feasible training and inference speeds. The gating network is trained alongside the experts using a load balancing loss to ensure even utilization and prevent any single expert from becoming a bottleneck.
Feature-wise Linear Modulation (FiLM) is integrated into the Transformer architecture to incorporate information regarding predicted Space Group Symmetry. This is achieved by conditioning each Transformer layer with a scaling and shifting parameter, derived from the predicted symmetry, applied to the layer’s feature maps. Specifically, for each feature map [latex]x_i[/latex], FiLM calculates [latex]y_i = \gamma_i * x_i + \beta_i[/latex], where [latex]\gamma_i[/latex] and [latex]\beta_i[/latex] are learned scaling and shifting parameters dependent on the predicted Space Group Symmetry. This conditioning mechanism allows the model to bias its generation process towards structurally valid configurations, effectively guiding the output towards solutions consistent with the identified symmetry constraints.
Constrained Beam Search is utilized to address the combinatorial challenge of Wyckoff position assignment within predicted crystal structures. Wyckoff positions define equivalent sites within a space group, and their correct assignment is crucial for structural validity. Instead of exhaustively searching all possible assignments – an operation with exponential complexity – Beam Search maintains a limited set of candidate solutions, iteratively expanding them based on a scoring function that prioritizes compatibility with predicted symmetry and minimizes structural violations. By pruning less promising candidates at each step, computational cost is significantly reduced while ensuring the generated structures adhere to the imposed space group symmetry constraints. This approach allows for efficient exploration of the assignment space and facilitates the generation of structurally plausible crystal structures.
Validating Integrity and Measuring Predictive Power
To guarantee the validity and physical realism of newly generated crystal structures, a rigorous rectification process is implemented. This involves both Coordinate Rectification and Lattice Rectification, critical steps that enforce adherence to predicted Wyckoff symmetry – a fundamental principle governing the arrangement of atoms within a crystal lattice. Coordinate Rectification adjusts the fractional coordinates of each atom, ensuring they conform to the symmetry constraints dictated by the space group. Simultaneously, Lattice Rectification refines the lattice parameters – the dimensions and angles defining the unit cell – to further align with the predicted symmetry. This dual-correction mechanism doesn’t merely ensure mathematical consistency; it actively promotes the creation of structurally sound and physically plausible materials, effectively filtering out unrealistic or unstable configurations before further evaluation.
Rigorous evaluation of the generated crystal structures relies on established metrics designed to quantify their quality and relevance. The research utilizes SUN Metrics – encompassing Stability, Uniqueness, and Novelty – to comprehensively assess each structure’s thermodynamic favorability, distinctiveness from existing materials, and potential for groundbreaking properties, respectively. Complementing this is the Matching Rate, a crucial indicator of similarity to known, experimentally verified compounds. This dual approach ensures that the framework doesn’t merely produce plausible structures, but rather, those with a high probability of real-world existence and potentially valuable characteristics, bridging the gap between computational prediction and materials discovery.
Evaluations reveal the Symmetry-Driven Generative Framework significantly outperforms existing methods in materials structure generation, achieving a remarkable 376% increase in the overall Stability, Uniqueness, and Novelty (SUN) metric when tested across the MP-20, MPTS-52, and Perov-5 datasets, compared to DiffCSP++. This improvement is driven by substantial gains in stability-124% on MP-20 and approximately 255% on MPTS-52-and a marked increase in novelty, reaching 71% on MP-20 and 53% on MPTS-52. Critically, this framework doesn’t compromise accuracy; it simultaneously achieves state-of-the-art Matching Rate performance, indicating a superior ability to generate plausible and previously unseen crystal structures with high fidelity.
Expanding the Horizon: Towards Predictive Materials Design
Current materials prediction often simplifies reality, overlooking crucial factors like temperature, pressure, and intricate chemical interactions. This framework, however, possesses the adaptability to integrate these complexities, moving beyond idealized models. By incorporating a broader range of chemical constraints – accounting for nuanced bonding preferences and stoichiometry – and environmental factors like thermal expansion or stress, the system can more accurately predict stable crystal structures under realistic conditions. This enhanced predictive power is achieved through refined algorithms that assess the energetic landscape of potential structures, weighting stability not just by intrinsic chemical bonds, but also by external influences. Consequently, researchers can anticipate material behavior in operating environments, accelerating the design of robust and functional materials tailored for specific applications – from high-performance alloys to energy storage devices.
The efficiency of materials discovery can be substantially improved by incorporating active learning strategies into computational workflows. Rather than passively screening numerous potential materials, these methods intelligently select the most promising candidates for further investigation – typically through simulations or experiments. This iterative process focuses computational resources on regions of chemical space likely to yield desired properties, effectively navigating the vast compositional landscape with greater speed and precision. By continuously refining its understanding based on newly acquired data, the system prioritizes exploration of areas with high potential, minimizing redundant calculations and accelerating the identification of novel materials tailored to specific applications. This approach represents a paradigm shift from exhaustive searching to targeted discovery, promising to unlock materials innovation at an unprecedented rate.
The predictive power of this materials design framework extends far beyond simple bulk structures, offering a pathway to understanding and controlling the nuanced properties dictated by defects and interfaces – critical elements often governing material performance. These imperfections, traditionally challenging to model accurately, significantly influence characteristics like strength, conductivity, and catalytic activity. By adapting the framework to specifically address atomic-scale irregularities and the complex interactions at material boundaries, researchers can move beyond theoretical estimations and directly predict how these features impact macroscopic behavior. This capability promises not only accelerated materials discovery, tailored to specific applications, but also a deeper fundamental understanding of material behavior, ultimately enabling the design of materials with unprecedented properties and functionalities.
The pursuit of rigorous crystal structure prediction, as detailed in this work, echoes a fundamental principle of elegant design. It isn’t about adding layers of complexity, but distilling the core geometric truths. As Albert Einstein once stated, “It can scarcely be said which is more incredible: the scope of his knowledge or his pedagogical tact.” This framework, by enforcing symmetry constraints during the diffusion process, embodies this sentiment. The generative model doesn’t simply create; it reveals inherent order, much like discovering an existing principle rather than inventing a new one. The focus on Wyckoff positions and space groups isn’t a limitation, but a refinement-a demonstration that true power lies in understanding and respecting fundamental constraints.
Where Do We Go From Here?
The present work, while demonstrating a marked improvement in guided materials discovery, merely clarifies the inevitable limitations of its approach. To infer symmetry is not to be symmetry. The framework excels at enforcing known constraints, yet remains tethered to the predictive capacity of the initial diffusion model. The true challenge, consistently evaded, is not refining the map, but abandoning the territory of approximation altogether. A generative process truly decoupled from initial guesswork – a process that defines stability, rather than seeking it – remains elusive.
Future iterations will undoubtedly focus on expanding the scope of detectable space groups and refining the geometric precision of Wyckoff position enforcement. However, such improvements represent diminishing returns. A more fruitful, though considerably more difficult, path lies in exploring the fundamental relationship between symmetry, information, and the very definition of a ‘stable’ material. The current paradigm assumes stability is a discoverable property; perhaps it is a construct, an artifact of the search itself.
Ultimately, the persistence of complexity within these models serves only as a testament to the limitations of the questions being asked. If a stable crystal structure cannot be described with elegant simplicity, then the fault lies not with the material, but with the inadequate language used to define it.
Original article: https://arxiv.org/pdf/2602.17176.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- Beyond Accuracy: Gauging Trust in Human-AI Teams
- Gold Rate Forecast
- Clash Royale Balance Changes March 2026 — All Buffs, Nerfs & Reworks
- The most surprising Hannah Montana cameos: From John Cena to Dwayne Johnson and even a Coronation Street soap star as show celebrates its 20th anniversary
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- EMEA Masters Winter 2026 introduces official Qualifier for Esports World Cup
- Genshin Impact Version 6.5 Leaks: List of Upcoming banners, Maps, Endgame updates and more
- Brawl Stars Sands of Time Brawl Pass brings Sandstalker Lily and Sultan Cordelius sets, along with chromas and more
- Total Football free codes and how to redeem them (March 2026)
2026-02-21 08:49