Author: Denis Avetisyan
Researchers have developed a powerful deep learning framework to decode the complex spatial arrangement of cells and their genomic interactions within tissues.
![CellScape constructs a cellular landscape by jointly modeling spatial proximity and gene co-expression, employing a dual-branch architecture that generates both spatial embeddings [latex]Z_{\text{spatial}}[/latex] and intrinsic gene expression embeddings [latex]Z_{\text{intrinsic}}[/latex], thereby enabling a nuanced understanding of cellular organization and facilitating diverse downstream analyses in spatial omics data.](https://arxiv.org/html/2602.12651v1/figs/fig1_v3.png)
CellScape integrates spatial transcriptomics and genomic data to reveal tissue architecture and improve multi-sample analysis through a novel dual-branch network and effective batch effect correction.
Dissecting tissue architecture requires bridging the gap between genomic identity and spatial context, yet existing computational methods struggle with the complexity of spatial transcriptomics data. To address this, we present CellScape, a deep learning framework detailed in ‘Uncovering spatial tissue domains and cell types in spatial omics through cross-scale profiling of cellular and genomic interactions’, which jointly models cellular interactions and genomic relationships to generate comprehensive tissue representations. This approach improves spatial domain segmentation and enables robust multi-sample analyses by effectively integrating spatial and genomic information. How might these cross-scale profiling methods refine our understanding of cellular organization and disease pathogenesis within complex tissues?
Beyond Averaging: The Illusion of Bulk Analysis
Conventional methods of gene expression analysis typically involve homogenizing tissue samples, effectively averaging the signals from numerous cells and their unique locations. This approach obscures the inherent spatial heterogeneity within tissues, where gene activity can vary dramatically even between neighboring cells. Consequently, critical information regarding cellular interactions, tissue organization, and the influence of the microenvironment is lost. The resulting ‘bulk’ expression data provides only a generalized overview, failing to capture the nuanced expression patterns that define distinct cell types, developmental stages, or disease states. This limitation hinders a complete understanding of complex biological processes, as gene expression is rarely uniform and is often tightly linked to a cell’s position and its communication with surrounding cells.
The conventional methods of gene expression analysis, while valuable, often treat tissues as a homogenous blend, effectively overlooking the intricate architecture and specialized roles of individual cells. This averaging process can conceal critical information about how cells organize themselves and express genes differently based on their location and interactions with neighboring cells. Consequently, researchers may miss nuanced changes in cellular phenotypes-the observable characteristics resulting from gene expression-that are vital for understanding both normal development and the progression of disease. For instance, subtle variations in gene expression within a tumor microenvironment, obscured by bulk analysis, can significantly impact therapeutic response and metastasis, highlighting the limitations of approaches that fail to capture spatial context. Ultimately, a more detailed understanding of gene expression at the single-cell level, within the tissue’s spatial framework, is essential for unlocking a complete picture of biological processes.
Accurate biological interpretation increasingly demands that gene expression be understood not as a uniform property of a tissue, but as a spatially resolved phenomenon. Traditional methods, by averaging signals across entire samples, lose vital information about how gene activity varies within the tissue’s architecture. This localized expression is crucial; cells in different positions experience unique microenvironments and interact with neighbors in ways that profoundly influence their behavior. Consequently, understanding which genes are active, and at what levels, in specific locations is essential for deciphering complex processes like embryonic development, immune responses, and cancer progression. Failing to account for this spatial dimension risks obscuring critical details about cellular phenotypes and interactions, ultimately hindering the ability to build comprehensive and accurate models of biological systems.
Mapping the Transcriptome in Space: A Toolkit of Technologies
Spatial transcriptomics technologies, including Visium, Slide-seqV2, STARmap PLUS, and Stereo-seq, enable the simultaneous measurement of gene expression and the retention of information regarding the original location of those transcripts within a tissue sample. Traditional RNA sequencing methods lose this spatial context during tissue processing; these technologies address this limitation by employing various techniques to link gene expression data to specific coordinates. This allows researchers to investigate how gene expression patterns vary across different regions of a tissue, providing insights into cellular organization, tissue architecture, and the functional roles of genes within a spatial framework. The resulting data facilitates the identification of spatially-defined cell types, the analysis of cell-cell interactions, and the characterization of localized gene expression programs.
Spatial transcriptomics technologies utilize distinct methodologies to spatially resolve gene expression. Microfabricated arrays, such as those used in Visium, employ spatially barcoded spots to capture mRNA, providing relatively low resolution but high throughput. Bead-based systems, exemplified by Slide-seqV2, leverage oligonucleotide-conjugated beads distributed across tissue sections; bead location defines spatial coordinates, achieving higher resolution than arrays. STARmap PLUS and Stereo-seq utilize DNA nanoballs containing spatially barcoded oligonucleotides, allowing for highly multiplexed and high-resolution mapping of transcripts; Stereo-seq further enhances resolution by sequencing transcripts in a spatially defined grid. The choice of methodology directly impacts the achievable resolution, ranging from approximately 55 μm for Visium to sub-cellular resolution with Stereo-seq.
Spatial transcriptomics technologies vary significantly in their operational parameters, demanding careful consideration during experimental design. Visium generally offers lower resolution but higher throughput and lower cost per sample, making it suitable for broad surveys. Slide-seqV2 provides improved resolution over Visium, albeit with a reduction in throughput and increased cost. STARmap PLUS and Stereo-seq achieve the highest resolutions, allowing for near-single-cell resolution, but at the expense of significantly lower throughput and substantially higher costs per sample. The optimal choice depends on the specific research question; experiments prioritizing large-scale tissue mapping may favor Visium, while those requiring precise localization of gene expression in specific cell types will likely necessitate the use of STARmap PLUS or Stereo-seq, despite the associated budgetary and logistical considerations.
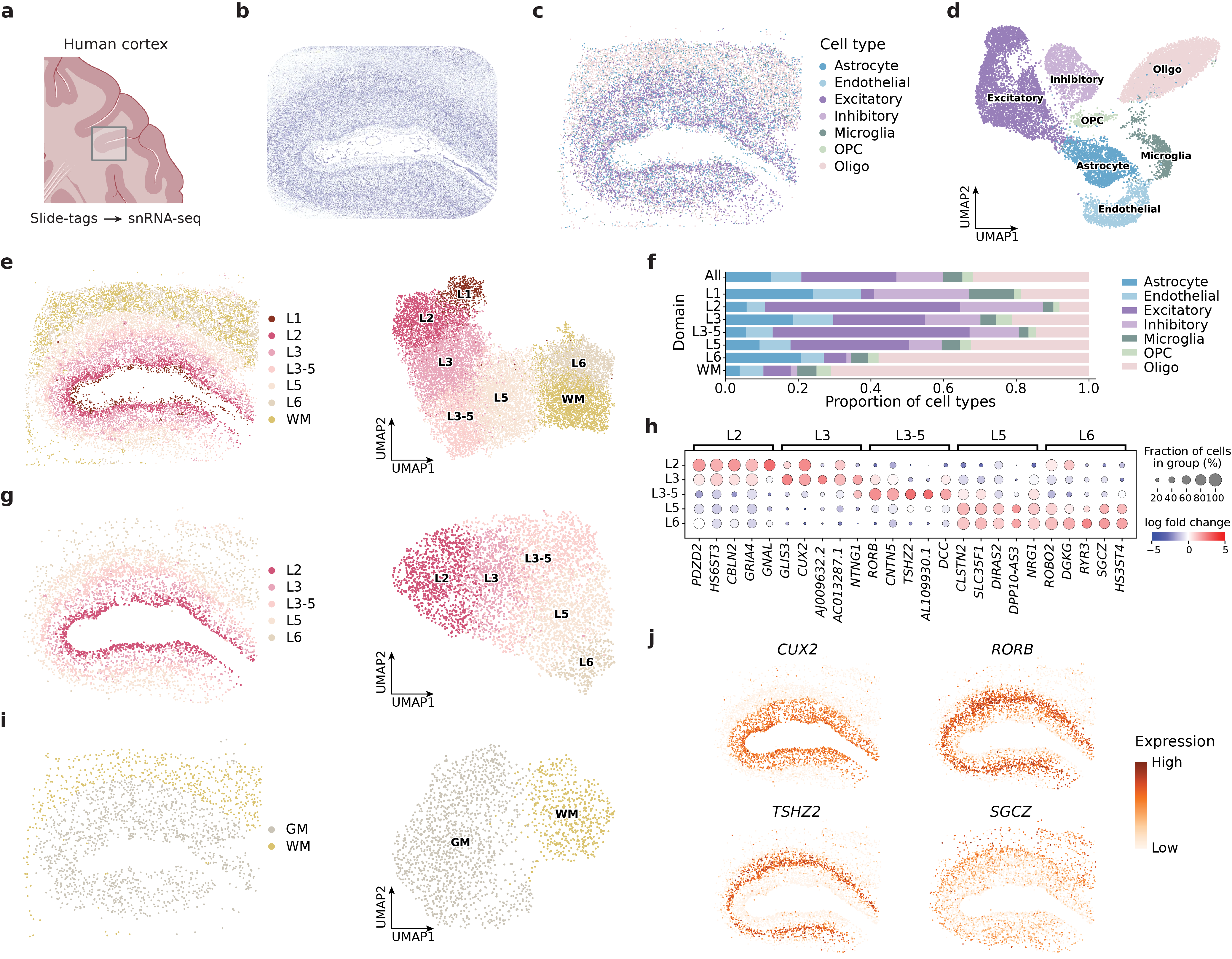
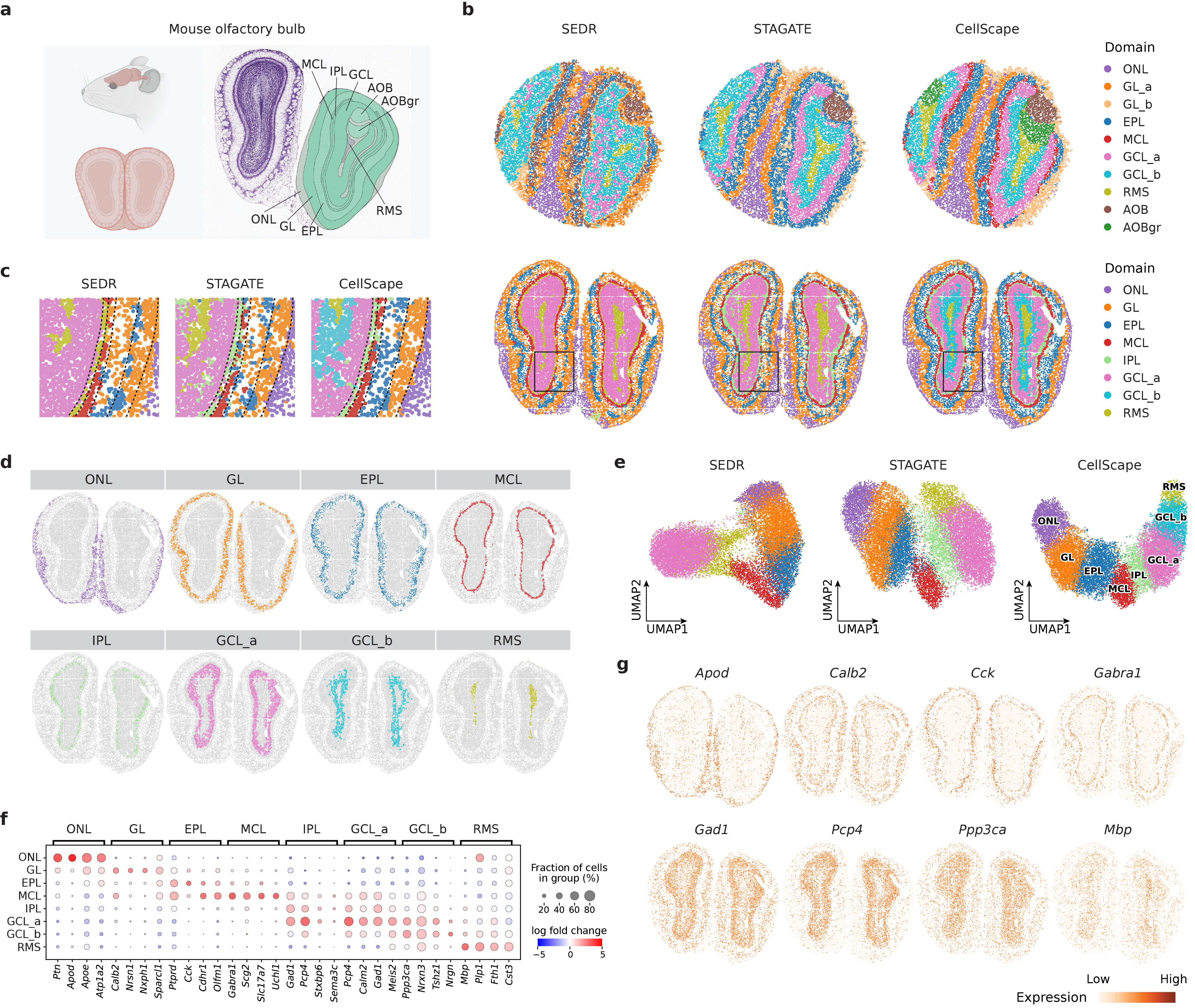
![CellScape accurately delineates spatial domains across multiple single-cell datasets-including osmFISH, 10x Genomics Visium, and STARmap-and demonstrates superior segmentation performance compared to baseline methods, as validated by manual annotations and quantitative metrics [latex] (p<0.001) [/latex], while also effectively integrating data across samples and slices as shown by UMAP visualizations of learned cell embeddings.](https://arxiv.org/html/2602.12651v1/figs/fig5.png)
CellScape: A Deep Learning Framework for Spatial Resolution
CellScape is a deep learning framework designed for the integrated analysis of spatial transcriptomics data, combining gene expression information with the physical location of cells within a tissue. The framework accepts as input both gene expression matrices and spatial coordinates, allowing for the simultaneous consideration of cellular phenotype and tissue organization. This integration is achieved through a multi-modal deep learning architecture, enabling the identification of spatially-defined cell types and the reconstruction of tissue structure based on gene expression patterns. By jointly analyzing genomic and spatial data, CellScape facilitates investigations into how cellular phenotypes are related to their microenvironment and contributes to a more comprehensive understanding of tissue biology.
CellScape utilizes dimensionality reduction via Autoencoders to efficiently process high-dimensional spatial transcriptomics data, transforming it into a lower-dimensional representation while preserving essential biological information. Beyond simple data compression, the framework models intercellular relationships using Graph Neural Networks (GNNs). These GNNs represent tissues as graphs, where cells are nodes and cell-cell interactions – inferred from transcriptomic similarity and spatial proximity – are edges. This graph-based approach allows CellScape to capture and analyze complex cellular communication patterns and spatial organization, enabling the identification of functionally related cell populations and their interactions within the tissue microenvironment.
CellScape’s performance is quantitatively assessed using metrics designed to evaluate both clustering quality and the preservation of spatial relationships within the analyzed tissue. Specifically, Normalized Mutual Information (NMI) quantifies the agreement between the identified clusters and known or expected cell type annotations, while Homogeneity measures the extent to which cells within each cluster exhibit consistent spatial localization. On the osmFISH dataset, CellScape achieved an NMI score of 0.745, demonstrating superior performance compared to other evaluated computational methods for spatial transcriptomics data analysis. This result indicates a greater ability to accurately identify and spatially organize cell types based on gene expression profiles.
![CellScape analysis of mouse brain tissue reveals disease-associated spatial and molecular alterations in Alzheimer’s disease, including shifts in cell-type composition, differential microglial gene expression ([latex]\Delta\Delta[/latex] representing AD versus control), and colocalization of DAM markers like Cst7 and Trem2 with [latex]A\beta[/latex] plaques.](https://arxiv.org/html/2602.12651v1/figs/fig3.png)
Unlocking Biological Insight: From Maps to Mechanisms
The intricate architecture of tissues isn’t simply a collection of cells, but a carefully orchestrated arrangement where cell type and location dictate function. Recent advances in spatial transcriptomics, particularly when coupled with computational tools like CellScape, now allow researchers to map gene expression patterns directly within the tissue context. This approach moves beyond bulk RNA sequencing, which averages signals across many cells, to pinpoint the precise location of different cell types and uncover how they interact with their neighbors. By identifying spatially defined cell populations, scientists can decipher the organizational principles governing tissue development, homeostasis, and disease progression – revealing, for instance, how immune cells cluster around tumor cells or how neuronal circuits are organized within the brain. This capability provides an unprecedented level of detail, fostering a deeper understanding of the complex relationship between a cell’s identity, its physical surroundings, and its ultimate role within the larger tissue ecosystem.
The ability to map gene expression patterns within the precise architecture of tissues opens new avenues for dissecting complex biological phenomena. Researchers can now investigate how cells orchestrate developmental programs by observing spatially defined changes in gene activity, revealing the intricate interplay between location and cellular fate. Similarly, disease mechanisms, such as tumor progression or inflammatory responses, are no longer viewed as uniform processes, but rather as spatially heterogeneous events with distinct microenvironments. This localized understanding is crucial for evaluating the efficacy of therapeutic interventions; researchers can determine if a drug is reaching the intended target cells and inducing the desired changes within specific tissue regions, offering a more nuanced and accurate assessment of treatment outcomes than traditional bulk analyses allow.
Evaluations reveal CellScape to be a robust and accurate tool for analyzing spatial transcriptomics data, consistently exceeding the performance of established methods across diverse datasets. Notably, when applied to the 10x Genomics Visium DLPFC dataset – a standard benchmark in the field – CellScape achieved a Homogeneity score of 0.659. This metric signifies a high degree of agreement between the cell type annotations predicted by the algorithm and those derived from established reference datasets, suggesting the method effectively captures the true biological organization of the tissue. Such consistent and reliable performance positions CellScape as a valuable asset for researchers aiming to decipher complex spatial gene expression patterns and uncover nuanced insights into tissue structure and function.
The field of spatial transcriptomics is poised for significant advancements, with ongoing research concentrating on three key areas: resolution, throughput, and analytical sophistication. Increasing resolution will allow scientists to pinpoint gene expression at the subcellular level, revealing nuanced details of cellular function and interaction. Simultaneously, improvements in throughput-the speed and scale of data acquisition-will enable the comprehensive profiling of larger and more complex tissues. Crucially, these technological leaps are coupled with the development of novel analytical methods, including machine learning algorithms, designed to interpret the vast datasets generated and translate them into biologically meaningful insights, ultimately promising a complete and dynamic understanding of the transcriptomic landscape within tissues and its role in both health and disease.
The pursuit of delineating tissue architecture through computational means inevitably reveals the inherent fragility of imposed order. CellScape, with its dual-branch network, attempts to map cellular interactions and gene expression, but this very act of mapping establishes a new dependency-a reliance on the algorithm’s interpretation of reality. As Claude Shannon observed, “The most important thing is to avoid being misled by the apparent simplicity of the problem.” This resonates deeply with the work; the seeming clarity of spatial domain segmentation masks the complex, often arbitrary, decisions embedded within the deep learning framework. The system grows, certainly, but not without accruing technical debt-the prophecy of future failure encoded in every architectural choice.
What Lies Ahead?
CellScape, like any attempt to map the interior of a living thing, offers a snapshot, not a photograph. The architecture it reveals is provisional, a momentary equilibrium in a system perpetually seeking new disequilibrium. The framework’s success in integrating spatial and genomic data should not be mistaken for comprehension. It merely refines the questions, reveals the elegance of the failures yet to come. A system that never misclassifies a cell type is, demonstrably, not interacting with a living tissue.
Future iterations will undoubtedly focus on scaling – larger tissues, more samples, higher resolution. But true progress lies not in diminishing error, but in embracing its inevitability. The real challenge isn’t correcting for batch effects, but understanding them as inherent properties of the biological process itself. Each ‘correction’ is a prophecy of the variation that will inevitably emerge, a narrowing of the space for adaptation.
The pursuit of perfect segmentation, of definitive cell type identification, is a seductive trap. Perfection leaves no room for people – or, in this case, for the plasticity of cells, the ambiguity of boundaries. The most fruitful avenues of research will likely explore not what is known about tissue organization, but what remains stubbornly, beautifully, unknowable.
Original article: https://arxiv.org/pdf/2602.12651.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Invincible Season 4 Episode 4 Release Date, Time, Where to Watch
- How Martin Clunes has been supported by TV power player wife Philippa Braithwaite and their anti-nepo baby daughter after escaping a ‘rotten marriage’
- Beyond Accuracy: Gauging Trust in Human-AI Teams
- Gold Rate Forecast
- Clash Royale Balance Changes March 2026 — All Buffs, Nerfs & Reworks
- CookieRun: OvenSmash coupon codes and how to use them (March 2026)
- Roco Kingdom: World China beta turns chaotic for unexpected semi-nudity as players run around undressed
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- Charlie Day Confirms What Always Sunny Scene Is His Career Highlight
- We talked to ‘Bachelorette’ Taylor Frankie Paul. Then reality hit pause on her TV career
2026-02-16 20:53