Author: Denis Avetisyan
Researchers have released a comprehensive dataset and evaluation platform to accelerate the development of AI tools that can determine a molecule’s structure from its NMR spectrum.
NMRGym provides a standardized, large-scale benchmark for advancing deep learning methods in molecular structure elucidation using nuclear magnetic resonance spectroscopy.
Despite the power of deep learning in automating molecular structure elucidation via Nuclear Magnetic Resonance (NMR) spectroscopy, progress is hindered by reliance on synthetic datasets that fail to capture real-world spectral diversity. To address this critical gap, we introduce ‘NMRGym: A Comprehensive Benchmark for Nuclear Magnetic Resonance Based Molecular Structure Elucidation’, a large-scale, standardized benchmark comprising [latex]269,999[/latex] unique molecules paired with high-fidelity [latex]\${^1}\$H[/latex] and [latex]\${^{13}}\$C[/latex] spectra derived from experimental data. This resource, coupled with a strict scaffold-based data split and comprehensive evaluation suite, establishes a new standard for fair comparison and rigorous assessment of structure elucidation methodologies. Will NMRGym accelerate the development of more robust and accurate deep learning models capable of deciphering complex molecular structures directly from experimental NMR data?
The Enduring Challenge of Molecular Definition
The cornerstone of chemical understanding rests upon the precise determination of molecular structure, yet achieving this is often a protracted and demanding process. Historically, chemists have relied on techniques like X-ray crystallography and meticulous analysis of spectroscopic data, each requiring significant expertise and considerable time investment. Even with advancements in analytical instrumentation, interpreting the resulting data-complex patterns of diffraction or absorption-demands a deep understanding of chemical principles and a skilled ability to connect observed phenomena to specific atomic arrangements. This reliance on expert interpretation creates a bottleneck in research, limiting the speed at which new compounds can be characterized and understood, and hindering progress in fields ranging from drug discovery to materials science. Consequently, the development of more automated and efficient methods for structure elucidation remains a central challenge in modern chemistry.
Nuclear Magnetic Resonance (NMR) spectroscopy remains a cornerstone of molecular structure determination, generating highly detailed information about the connectivity and environment of atoms within a molecule. However, translating this wealth of data into a definitive structural assignment is often a significant challenge, particularly as molecular complexity increases. Manual spectral analysis demands considerable expertise and time, requiring chemists to painstakingly interpret subtle patterns and correlations within multi-dimensional spectra. The inherent complexity arises from signal overlap, dynamic processes, and the sheer volume of data produced by modern NMR experiments, creating a substantial bottleneck in research workflows and hindering the rapid characterization of novel compounds. This limitation is especially pronounced in fields like natural product chemistry and metabolomics, where samples often contain a multitude of closely related molecules.
Automated structure elucidation, a promising avenue for accelerating chemical discovery, currently faces significant limitations due to the incomplete nature of existing nuclear magnetic resonance (NMR) databases. While resources like NMRShiftDB represent valuable efforts to catalog predicted and experimental chemical shifts, their coverage remains far from comprehensive, particularly for complex organic molecules, natural products, and those containing uncommon functional groups or isotopes. This restricted data availability directly impedes the training and validation of machine learning algorithms designed to predict chemical shifts and, consequently, to deduce molecular structures from spectral data. The scarcity of reference data forces reliance on less accurate estimations or necessitates extensive, time-consuming manual analysis, effectively negating the potential benefits of automation and hindering progress in fields such as drug discovery and materials science. Expanding these databases with experimentally verified data, alongside standardized data formats and improved accessibility, is therefore crucial to unlocking the full potential of computational approaches to molecular structure determination.
Progress in computational chemistry, particularly in automated structure elucidation, is significantly hampered by a critical lack of robust, publicly available datasets. While algorithms can be designed to predict or interpret spectroscopic data, their efficacy remains largely unproven without comprehensive benchmarks for training and validation. Existing datasets are often fragmented, inconsistent in their standardization, or limited in the diversity of chemical structures represented. This scarcity prevents researchers from objectively assessing the performance of new computational methods, identifying their limitations, and refining them for broader applicability. Consequently, the field relies heavily on expert interpretation and manual analysis, hindering the potential for rapid, accurate, and scalable molecular structure determination – a cornerstone of modern chemical research and drug discovery.
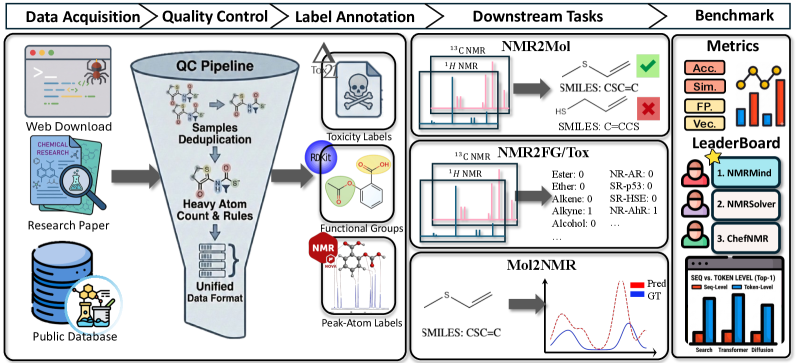
A Foundation for Data-Driven Discovery: Introducing NMRGym
NMRGym provides a benchmark dataset consisting of 269,999 paired molecular structures and corresponding experimental Nuclear Magnetic Resonance (NMR) spectra. This dataset represents a significant increase in scale compared to previously available resources for computational NMR, and is intended to serve as a standardized reference for evaluating and comparing predictive models. The dataset includes a variety of organic molecules, covering a broad chemical space, and is formatted to facilitate automated analysis and integration with machine learning workflows. Its size and comprehensive nature establish a new baseline for assessing the performance of algorithms designed for spectral prediction and structure elucidation in NMR spectroscopy.
Scaffold splitting was employed to create training, validation, and test sets within the NMRGym dataset, mitigating the risk of artificially inflated performance metrics due to model overfitting. This method divides molecules based on their underlying molecular scaffolds – the core structure excluding variable substituents – ensuring that no structurally similar compounds appear in multiple splits. Specifically, molecules sharing greater than 60% similarity in their scaffold representation, as determined by the Murcko scaffold, were assigned to different sets. This stringent separation provides a more realistic assessment of a model’s ability to generalize to novel chemical structures, as it prevents the model from simply memorizing patterns within closely related compounds and ensures a robust evaluation of its predictive capabilities.
The establishment of NMRGym as a standardized benchmark addresses a critical need for objective comparison of computational methods used in NMR spectral prediction and analysis. Prior to NMRGym, evaluating and comparing different algorithms was hampered by the lack of a universally accessible, consistently generated dataset. NMRGym provides a single, large-scale dataset against which the performance of various methods – including those employing machine learning and physics-based simulations – can be quantitatively assessed. This facilitates reproducible research, allows for clear identification of algorithmic strengths and weaknesses, and enables the tracking of incremental improvements in the field over time, ultimately accelerating the development of more accurate and efficient NMR analysis tools.
NMRGym supports the creation of automated workflows for determining molecular structures from NMR spectra by providing a substantial and standardized dataset for training and validating algorithms. Traditionally, structure elucidation relies heavily on expert manual interpretation of spectral data, a process that is both time-consuming and subject to human error. By offering nearly 300,000 molecule-spectrum pairs, NMRGym enables the development and benchmarking of machine learning models capable of predicting molecular structures directly from spectral input, potentially accelerating the pace of chemical discovery and reducing the need for extensive manual analysis. This automation extends to tasks such as peak picking, assignment, and connectivity determination, streamlining the entire structure elucidation process.
Shifting Paradigms: Machine Learning Approaches to Spectral Interpretation
Diffusion models and Transformer architectures represent a shift in computational approaches to structure elucidation by moving beyond traditional rule-based or template-matching methods. Diffusion models, originally developed for image generation, are adapted to generate realistic spectra from molecular structures through a process of iteratively adding and removing noise. Transformer architectures, known for their success in natural language processing, excel at identifying complex relationships within data; in this context, they analyze spectral patterns to infer underlying molecular connectivity. These models demonstrate the capacity to not only predict spectral features from known structures, but also to reverse the process – proposing plausible molecular structures consistent with observed spectra – thereby automating aspects of spectral interpretation and accelerating the structure elucidation process.
Machine learning models, specifically diffusion models and transformer architectures, are trained using the extensive dataset provided by NMRGym to establish correlations between molecular structures and their corresponding spectral features. This approach utilizes the large volume of simulated NMR data within NMRGym, enabling the models to learn complex relationships that would be difficult to define through traditional rule-based methods. The dataset encompasses a diverse range of molecules and associated spectra, allowing the models to generalize and accurately predict spectral characteristics from given molecular structures, or conversely, infer structural information from spectral data. This data-driven learning process circumvents the need for explicit programming of spectroscopic rules, instead relying on statistical patterns identified within the NMRGym dataset.
Heteronuclear Single Quantum Coherence (HSQC) and Correlation Spectroscopy (COSY) are two-dimensional Nuclear Magnetic Resonance (NMR) techniques that significantly aid in structure elucidation by providing connectivity information beyond that obtainable from one-dimensional 1H NMR. HSQC experiments establish direct one-bond 1H to 13C correlations, allowing for the unambiguous assignment of protonated carbons. COSY, conversely, reveals proton-proton couplings through scalar (J-coupling) and, less commonly, through-space interactions (Nuclear Overhauser Effect, NOE). By identifying which protons are directly bonded or in close proximity, COSY establishes spin systems, which are essential for piecing together the molecular framework. The combined use of HSQC and COSY enables a more complete and accurate determination of molecular connectivity, thereby streamlining the structure elucidation process.
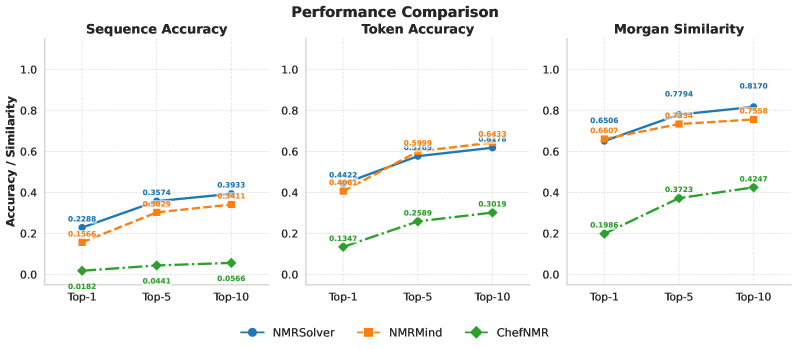
Evaluations using the NMRGym dataset demonstrate the performance of different machine learning approaches to spectral interpretation. Specifically, NMR-Solver achieved a Top-1 Sequence Accuracy of 22.88% in predicting molecular sequences from NMR data. This result represents a quantifiable improvement over the 15.66% Top-1 Sequence Accuracy achieved by NMRMind under the same testing conditions. These accuracy metrics provide a direct comparison of the algorithms’ ability to correctly identify molecular structures based on spectral input, highlighting NMR-Solver’s enhanced predictive capability within the NMRGym framework.
![Structural elucidation results were categorized into three scenarios-accurate identification, high-similarity deviations, and low-similarity failures-based on Tanimoto Similarity ([latex]Sim[/latex]) calculated using Morgan fingerprints.](https://arxiv.org/html/2601.15763v1/x5.png)
Unlocking Predictive Power: From Spectra to Molecular Properties
Recent advances demonstrate the feasibility of predicting molecular toxicity using machine learning models trained directly on Nuclear Magnetic Resonance (NMR) spectra. Algorithms like XGBoost and Random Forest excel at identifying complex relationships within spectral data, correlating specific NMR features with known toxicological outcomes. This approach bypasses the need for extensive computational chemistry or detailed molecular descriptors, offering a significantly faster and more efficient route to toxicity assessment. By learning from the unique ‘fingerprint’ of each molecule as revealed by its NMR spectrum, these models can rapidly screen large chemical libraries, prioritizing compounds with low predicted toxicity for further investigation in drug discovery and materials science. The ability to predict toxicity directly from spectral data represents a paradigm shift, accelerating the development of safer and more effective products.
The ability to predict molecular properties directly from Nuclear Magnetic Resonance (NMR) spectra represents a significant advancement for both drug discovery and materials science. Traditionally, determining these properties required extensive experimentation or computationally expensive simulations; however, machine learning models now offer a pathway for rapid, virtual screening of countless compounds. This accelerated process allows researchers to identify promising candidates with desired characteristics – such as specific biological activity or material strength – far more efficiently. By bypassing lengthy physical tests during initial stages, the time and resources required to develop new drugs and materials are substantially reduced, ultimately fostering innovation and accelerating the pace of scientific progress. This predictive capability isn’t merely about speed; it also unlocks the potential to explore a wider chemical space and discover compounds with previously unattainable properties.
The convergence of spectral prediction and property prediction represents a significant advancement in molecular design methodologies. By accurately forecasting both how a molecule will interact with electromagnetic radiation – its spectral signature – and what inherent characteristics it possesses, researchers can bypass traditional, often laborious, synthesis and testing phases. This integrated approach enables in silico screening of vast chemical spaces, identifying compounds likely to exhibit desired traits – such as specific bioactivity or material performance – before any physical experimentation occurs. Consequently, the platform facilitates a cycle of predictive modeling and targeted synthesis, drastically accelerating the discovery process and allowing for the rational creation of molecules optimized for specific applications. This capability promises to reshape fields ranging from pharmaceutical development to advanced materials science, fostering innovation through data-driven molecular engineering.
Recent advances in applying machine learning to nuclear magnetic resonance (NMR) spectroscopy have yielded promising results in molecular property prediction. Utilizing the comprehensive NMRGym dataset, the NMR-Solver model demonstrated superior performance, achieving a Morgan Tanimoto Similarity of 0.78 – establishing it as the top-performing model and exceeding the score of 0.73 attained by NMRMind. Furthermore, NMRFormer showcased significant capabilities in identifying functional groups, evidenced by its Macro F1-Score of 55.44%. These benchmarks highlight a considerable leap in the field, suggesting that automated analysis of NMR spectra can effectively discern molecular structure and, consequently, predict crucial characteristics for applications spanning drug development and materials science.
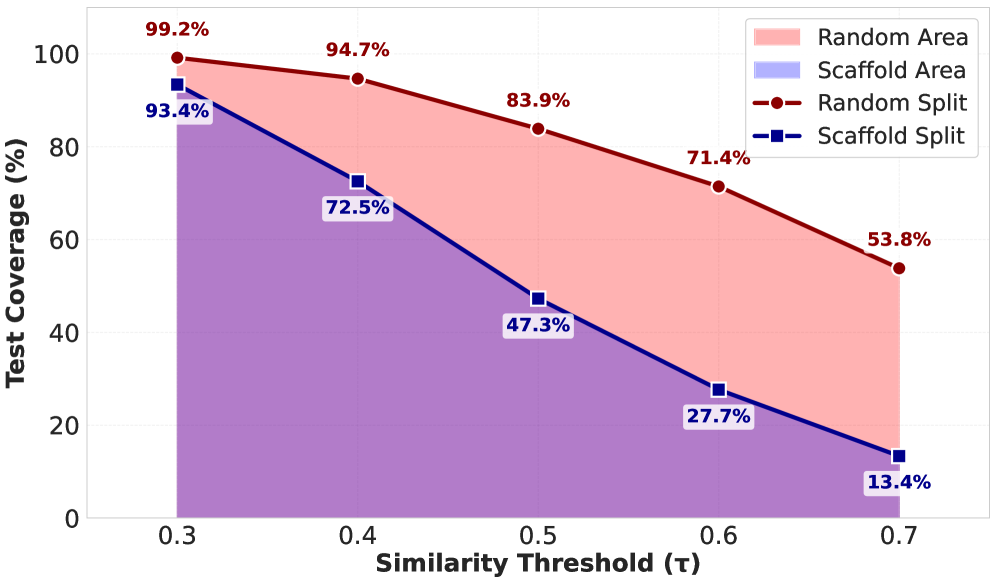
The pursuit of automated molecular structure elucidation, as detailed in this work, benefits from rigorous standardization. NMRGym offers precisely that – a controlled environment for evaluating algorithmic progress. Vinton Cerf observed, “The Internet treats everyone the same.” This echoes the intent behind the benchmark; by providing a level playing field, NMRGym allows for meaningful comparison of deep learning methods. Scaffold splitting, a core component of the dataset’s design, demands algorithms move beyond pattern matching toward genuine structural understanding. The simplicity of the benchmark’s goal – accurate prediction – belies the complexity of the underlying challenge, and a clear metric is paramount.
Where Do We Go From Here?
The creation of NMRGym represents, at its core, a formalized admission of difficulty. A system that requires a benchmark has already, implicitly, failed to achieve self-evident success. The value, then, lies not in demonstrating progress – any algorithm can be tailored to a specific dataset – but in exposing the remaining chasms in understanding. The true test will be performance on spectra demonstrably unlike those curated for this benchmark; spectra derived from molecules operating outside the comfortable confines of the training set.
Further refinement will inevitably focus on the nuances of spectral interpretation. The current paradigm often treats spectra as monolithic inputs, ignoring the inherent structural relationships that a human spectroscopist intuitively grasps. The next iteration must move beyond mere pattern recognition and towards a system capable of reasoning about molecular connectivity, chemical shifts, and coupling constants – a system that doesn’t simply predict, but explains.
Ultimately, the aim should be to diminish the need for such benchmarks entirely. A truly robust method would generalize beyond any specific dataset, deriving structural information directly from first principles. Until then, the cycle of creating increasingly elaborate benchmarks will continue – a testament not to advancement, but to the enduring complexity of the problem, and the humility required to acknowledge it.
Original article: https://arxiv.org/pdf/2601.15763.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Invincible Season 4 Episode 4 Release Date, Time, Where to Watch
- Physics Proved by AI: A New Era for Automated Reasoning
- How Martin Clunes has been supported by TV power player wife Philippa Braithwaite and their anti-nepo baby daughter after escaping a ‘rotten marriage’
- CookieRun: OvenSmash coupon codes and how to use them (March 2026)
- Total Football free codes and how to redeem them (March 2026)
- Invincible Creator on Why More Spin-offs Haven’t Happened Yet
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- Goddess of Victory: NIKKE 2×2 LOVE Mini Game: How to Play, Rewards, and other details
- Gold Rate Forecast
- American Idol vet Caleb Flynn in solitary confinement after being charged for allegedly murdering wife
2026-01-25 18:30