Author: Denis Avetisyan
A new approach leverages the power of artificial intelligence to directly interpret raw single-cell data, opening doors to faster and more accurate biological discovery.
Researchers introduce Omics-Native Reasoning, enabling Large Language Models to perform automated single-cell analysis and articulate biological hypotheses with improved accuracy and interpretability.
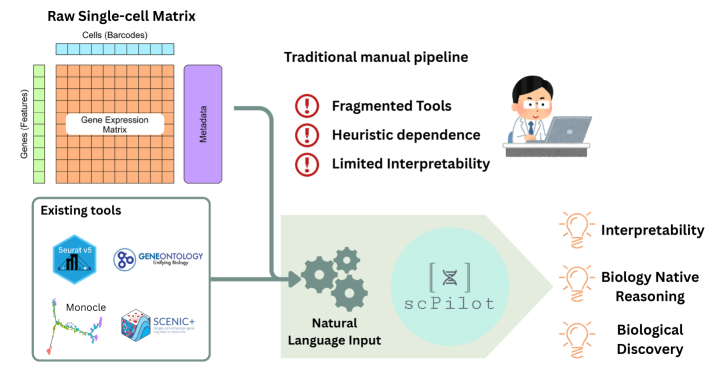
Despite the increasing volume of single-cell RNA sequencing data, extracting biologically meaningful insights remains a significant challenge due to the complexity of analysis and interpretation. This is addressed in ‘scPilot: Large Language Model Reasoning Toward Automated Single-Cell Analysis and Discovery’, which introduces Omics-Native Reasoning (ONR)-a framework enabling large language models to directly analyze raw omics data and articulate reasoning for tasks like cell-type annotation and trajectory inference. Experiments demonstrate that grounding LLMs in single-cell data substantially improves analytical accuracy and interpretability, with gains of up to 30% in trajectory reconstruction. Could this paradigm shift unlock a new era of auditable, diagnostically informative, and fully automated single-cell analyses?
Beyond Expression: Unraveling Biological Complexity with Reasoning
The advent of single-cell RNA sequencing (scRNA-seq) has unleashed an unprecedented volume of molecular data, yet simply identifying genes that exhibit differing levels of expression between cell populations proves insufficient for truly understanding the underlying biology. While differential gene expression analysis can pinpoint potential biomarkers, it often fails to capture the complex interplay of regulatory networks and cellular states. A list of altered genes, in isolation, doesn’t reveal why cells behave differently, nor does it elucidate the functional consequences of those changes. Meaningful interpretation demands moving beyond descriptive statistics to infer causal relationships, predict cellular responses, and ultimately, construct a comprehensive model of the biological system under investigation – a task requiring more than just identifying which genes are ‘up’ or ‘down’.
Conventional analyses of single-cell RNA sequencing data frequently fall short when attempting to decipher the intricate web of interactions governing cellular behavior. While identifying genes that are simply more or less active provides a starting point, these approaches often overlook the subtle regulatory relationships that define cellular states and responses. Cells rarely operate in isolation; instead, they communicate and influence each other through complex signaling pathways and feedback loops. Traditional statistical methods, geared towards detecting broad differences in gene expression, struggle to deconvolve these interwoven signals, potentially obscuring critical regulatory factors and misrepresenting the true dynamics at play. Consequently, researchers may miss vital insights into disease mechanisms, developmental processes, or the impact of environmental stimuli, highlighting the need for more sophisticated analytical frameworks.
The burgeoning field of single-cell RNA sequencing generates data-rich profiles of individual cells, yet simply identifying genes that exhibit differing levels of expression proves insufficient for a comprehensive understanding of biological systems. A transition is therefore necessary, moving beyond purely statistical analyses toward computational reasoning techniques that can model the complex interplay between genes, proteins, and cellular processes. This approach utilizes algorithms and knowledge-based systems to infer regulatory networks, predict cellular behavior, and ultimately, translate raw data into meaningful biological insights. By embracing computational reasoning, researchers can move past correlation to establish causation, enabling a deeper comprehension of cellular function and disease mechanisms than traditional methods allow.
scPilot: An Architecture for Omics-Native Reasoning
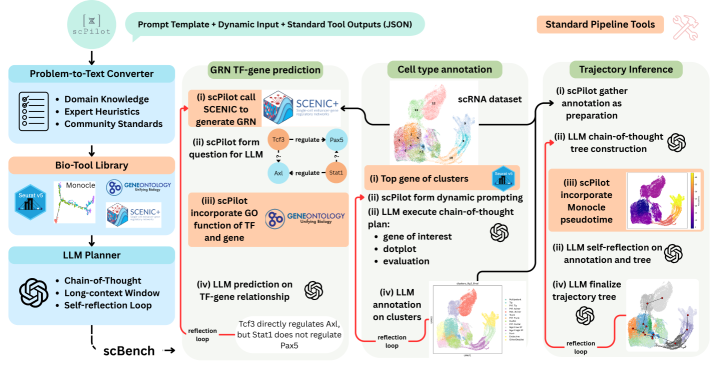
scPilot is a newly developed computational framework designed to implement Omics-Native Reasoning (ONR). This approach uniquely enables Large Language Models (LLMs) to directly process and interpret raw omics data, such as gene expression matrices and single-cell data, without requiring pre-processing or transformation into intermediate representations. By bypassing traditional data abstraction layers, scPilot facilitates a more granular and contextually rich analysis, allowing LLMs to leverage the full dimensionality of omics datasets. The framework establishes a direct interface between LLMs and raw data formats commonly used in genomics, proteomics, and other omics fields, effectively extending the reasoning capabilities of LLMs to the realm of biological data analysis.
scPilot enables complex single-cell analyses, including trajectory inference and gene regulatory network prediction, through the integration of Large Language Model (LLM) reasoning with external tool invocation. Rather than adhering to established analytical pipelines, scPilot dynamically utilizes specialized tools – such as those for differential expression, pseudotime calculation, and network analysis – based on the LLM’s interpreted analytical needs. This approach allows the LLM to chain together tool executions, processing outputs from one tool as inputs to others, and ultimately generating results without the constraints of pre-defined workflows. The LLM directs the analytical process, selecting and configuring tools as necessary, and interpreting the results to refine subsequent steps.
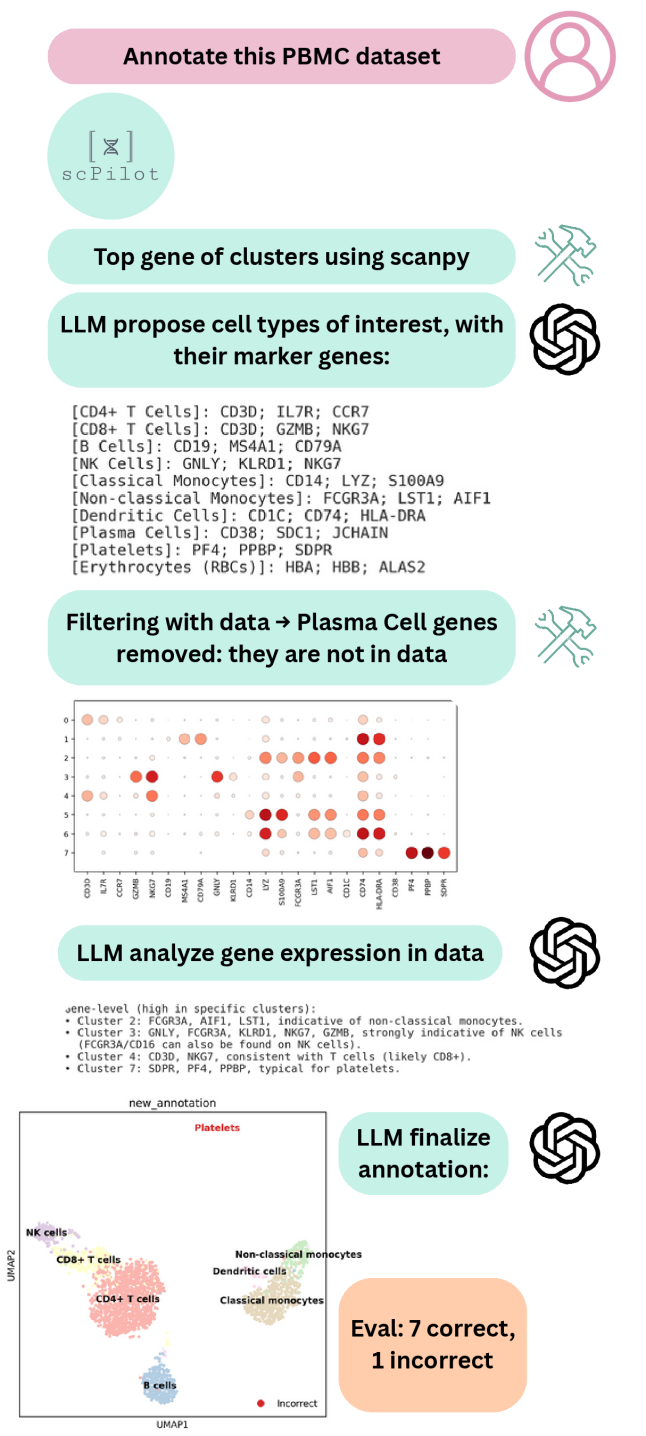
The scPilot framework enables transparent hypothesis generation and iterative analysis of single-cell data by allowing direct interaction with raw omics information. This contrasts with traditional pipeline-driven approaches, offering users greater control and visibility into the analytical process. Benchmarking demonstrates an 11% improvement in cell-type annotation accuracy when utilizing scPilot compared to existing single-cell analysis methods, suggesting increased precision in data interpretation facilitated by this exploratory approach.
Grounding Insights: Integrating Biological Knowledge
scPilot incorporates external biological knowledge through resources such as the Gene Ontology (GO) Database and Cell Ontology. The GO Database provides a standardized vocabulary describing gene functions in terms of biological processes, cellular components, and molecular functions, enabling functional annotation of predicted gene regulatory relationships. Cell Ontology defines cell types and their hierarchical organization, allowing scPilot to contextualize predictions within specific cellular lineages. By leveraging these ontologies, scPilot can assess the biological relevance of inferred interactions and prioritize interpretations based on established biological principles, thereby increasing the validity of its results.
scPilot employs a hypothesis prioritization strategy based on cross-referencing predicted gene regulatory relationships with established functional annotations from resources like the Gene Ontology (GO). This process assesses the biological relevance of each predicted interaction by determining if the involved genes share known functional associations – such as participation in the same biological process, molecular function, or cellular component. Predictions supported by these existing annotations receive higher prioritization scores, effectively filtering for hypotheses consistent with established biological knowledge and reducing the likelihood of spurious or biologically implausible results. This annotation-based filtering contributes to improved accuracy and interpretability of scPilot’s analyses.
The incorporation of external knowledge resources into scPilot’s analytical framework demonstrably improves the reliability and clarity of single-cell trajectory inference. By contextualizing predicted gene regulatory relationships with established functional annotations, scPilot moves beyond identifying simple correlations to generating biologically informed hypotheses. This knowledge integration is quantitatively supported by a 26% reduction in trajectory graph-edit distance compared to analyses performed without external validation, indicating improved concordance between predicted and established developmental or differentiation pathways.
Validation and the Mitigation of Reasoning Errors
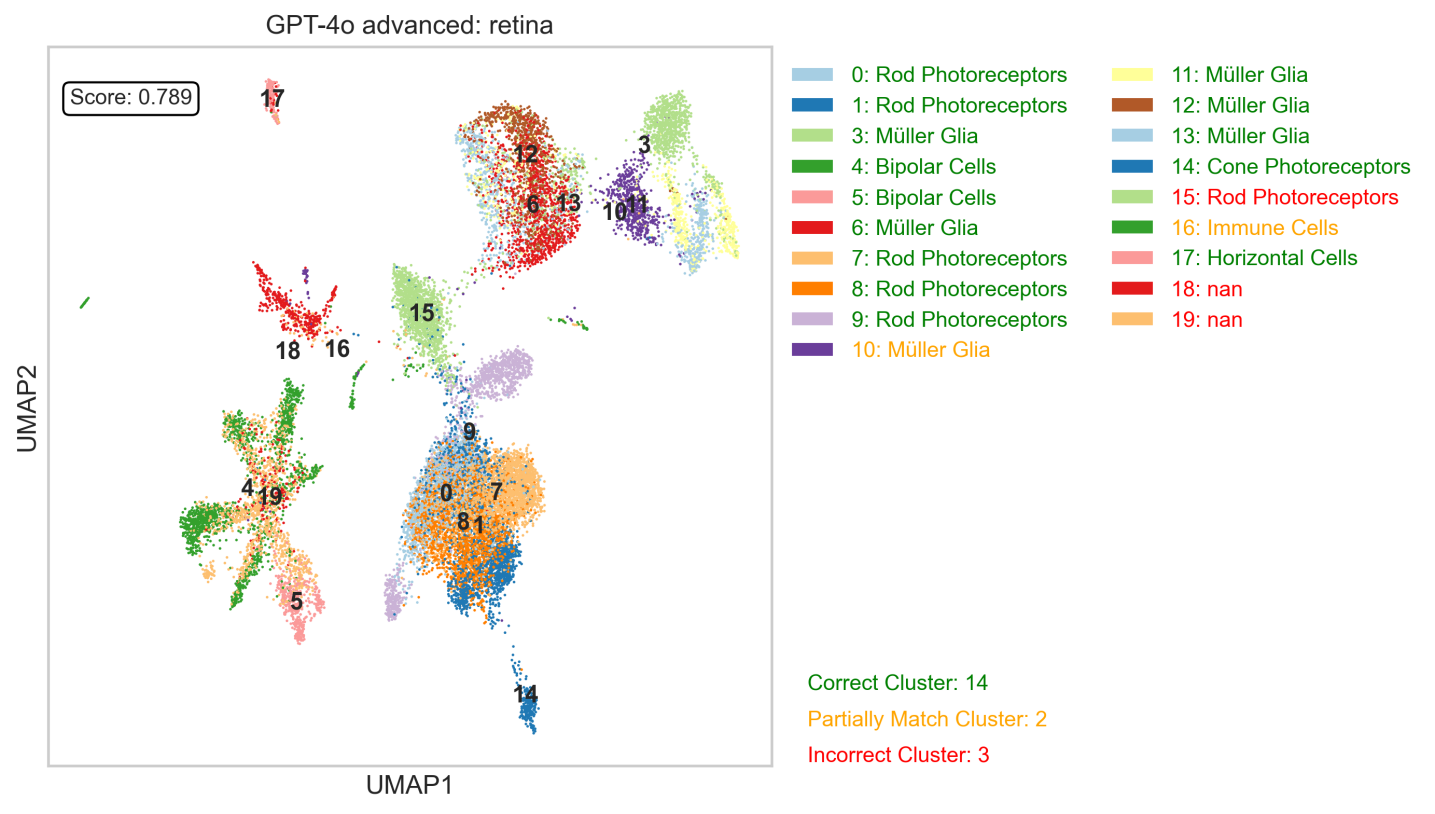
The scBench benchmark represents a significant step forward in assessing the capabilities of large language models applied to single-cell omics data, such as those utilized in scPilot. Unlike traditional metrics focused solely on numerical precision, scBench uniquely evaluates responses based on both quantitative accuracy and biological plausibility. This dual assessment is crucial; a model can generate numerically correct answers that are nonetheless meaningless in a biological context, or conversely, produce approximations that align with established biological principles. By scoring performance across these two dimensions, scBench provides a more holistic and reliable measure of an LLM’s reasoning abilities when interpreting complex omics datasets, ultimately fostering the development of tools that are both precise and biologically relevant.
Large language models, while powerful, are susceptible to generating “hallucinations” – outputs that appear plausible but are factually incorrect or lack biological coherence. This presents a significant hurdle in omics data analysis, where even subtle inaccuracies can lead to misleading conclusions and flawed hypotheses. These hallucinations aren’t simply random errors; they often stem from the LLM’s attempt to extrapolate beyond its training data or to fill gaps in knowledge with statistically likely, yet biologically improbable, information. Mitigating this risk requires not only robust benchmarking, like that offered by scBench, but also the development of validation strategies that specifically assess the biological validity of LLM-generated outputs, ensuring that the model’s reasoning aligns with established scientific principles and empirical evidence.
Establishing confidence in large language model (LLM) applications within biological research demands meticulous benchmarking and validation procedures. Without these, the potential for inaccurate interpretations and misleading conclusions significantly increases. Recent studies highlight the importance of this approach, demonstrating a 0.03 improvement in Area Under the Receiver Operating Characteristic curve (AUROC) for gene regulatory network (GRN) prediction when utilizing validated LLM-driven analyses compared to traditional methods. This seemingly small numerical gain underscores a substantial leap in predictive accuracy and reliability, suggesting that rigorous testing not only identifies potential errors but also refines model performance, ultimately paving the way for responsible and impactful integration of LLMs into the field of biological discovery.

The pursuit of Omics-Native Reasoning, as detailed in this work, necessitates a systemic approach to scientific AI. The model’s ability to directly interpret raw single-cell data, rather than relying on pre-processed summaries, echoes a holistic design philosophy. Vinton Cerf aptly stated, “The internet is not about technology; it’s about people.” Similarly, this research demonstrates that effective scientific AI isn’t merely about algorithms, but about enabling a deeper understanding of the underlying biological systems. The architecture proposed prioritizes interpretability, recognizing that a complex system’s behavior is dictated by its structure, and any attempt to refine one component requires consideration of the whole. Good architecture is invisible until it breaks, and only then is the true cost of decisions visible.
Looking Ahead
The pursuit of automated scientific discovery, as demonstrated by scPilot and its Omics-Native Reasoning approach, reveals a fundamental truth: the bottleneck is not computational power, but conceptual clarity. Scaling analyses-trajectory inference, gene network reconstruction-demands a scaling of understanding. The current paradigm often treats single-cell data as a solved problem, merely awaiting more sophisticated algorithms; instead, it appears the data is patiently awaiting a coherent question. The true leverage lies in framing analyses not as statistical exercises, but as logical deductions from first principles.
A critical limitation remains the reliance on pre-existing biological knowledge embedded within the Large Language Model. While impressive, this is fundamentally a sophisticated form of pattern matching. The next iteration must move beyond correlation and toward causal inference-a system capable of not just describing what is happening in the cell, but why. This necessitates integrating mechanistic models and experimental design directly into the reasoning process, turning the LLM from a knowledgeable observer into a proactive experimentalist.
Ultimately, the ecosystem of scientific AI will be judged not by benchmark scores, but by its ability to generate genuinely novel and testable hypotheses. The clarity of the underlying logic-the structure dictating behavior-will determine whether these systems remain elegant curiosities or become indispensable tools for unraveling the complexities of life.
Original article: https://arxiv.org/pdf/2602.11609.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Invincible Season 4 Episode 4 Release Date, Time, Where to Watch
- How Martin Clunes has been supported by TV power player wife Philippa Braithwaite and their anti-nepo baby daughter after escaping a ‘rotten marriage’
- Gold Rate Forecast
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- CookieRun: OvenSmash coupon codes and how to use them (March 2026)
- American Idol vet Caleb Flynn in solitary confinement after being charged for allegedly murdering wife
- Invincible Creator on Why More Spin-offs Haven’t Happened Yet
- Clash Royale Balance Changes March 2026 — All Buffs, Nerfs & Reworks
- Only One Straw Hat Hasn’t Been Introduced In Netflix’s Live-Action One Piece
- Roco Kingdom: World China beta turns chaotic for unexpected semi-nudity as players run around undressed
2026-02-16 04:05