Author: Denis Avetisyan
A new approach combines path sampling with artificial intelligence to automatically identify key molecular pathways and dramatically speed up the study of infrequent, yet crucial, events.
This work introduces AIMMD, a method leveraging machine learning to discover reaction coordinates and enhance the efficiency of transition path sampling for simulating rare molecular events.
Simulating rare events in molecular systems remains a persistent challenge due to the vast energy landscapes involved. The study ‘Path Sampling for Rare Events Boosted by Machine Learning’ introduces Artificial Intelligence for Molecular Mechanism Discovery (AIMMD), a novel framework integrating machine learning with transition path sampling to accelerate the identification of crucial reaction coordinates and efficiently explore these complex pathways. AIMMD achieves this by enabling on-the-fly estimation of committor probabilities, offering a robust and interpretable approach to understanding molecular dynamics. Will this automated discovery of reaction coordinates usher in a new era of mechanistic insight for previously intractable molecular processes?
The Challenge of Rare Events: A Fundamental Limitation
Standard molecular dynamics simulations, while powerful tools for investigating atomic behavior, face a fundamental hurdle when tasked with modeling infrequent occurrences. These simulations operate by numerically solving the equations of motion for all atoms in a system, effectively charting a trajectory through its possible configurations; however, rare events, by their very nature, occupy only a tiny fraction of this vast configuration space. Consequently, even extremely long simulations may fail to encounter these crucial states, leading to inaccurate or incomplete results. This insufficient sampling isn’t a matter of computational power, but rather a statistical limitation; the timescale for observing a rare event might far exceed the accessible simulation time. As a result, understanding phenomena like protein folding, ligand binding, or chemical reactions – all characterized by infrequent transitions – demands innovative approaches to overcome this inherent sampling problem.
The fundamental processes driving life and matter – from the intricate choreography of protein folding to the subtle shifts in chemical reactions – often involve events that occur on timescales or with probabilities inaccessible to standard computational methods. While these infrequent occurrences dictate the behavior of complex systems, their rarity presents a significant challenge; simulating them requires exploring vast configuration spaces, an endeavor quickly limited by computational resources. Consequently, despite advances in molecular dynamics, accurately modeling these crucial events remains largely intractable, hindering progress in fields ranging from drug discovery to materials science. Understanding the mechanisms governing these rare events necessitates innovative approaches capable of efficiently sampling the free energy landscape and uncovering the pathways that connect stable states.
Computational approaches to understanding infrequent molecular events necessitate techniques capable of efficiently mapping the free energy landscape, a complex representation of potential states and the energetic barriers between them. These methods move beyond simple trajectory calculations, instead focusing on identifying the minimum energy paths – transition pathways – that connect stable states. By strategically sampling configurations along these pathways, researchers can bypass the need for exhaustive simulations and directly assess the rates and mechanisms of rare events like protein conformational changes or chemical reactions. Advanced techniques, such as transition path sampling and umbrella sampling, achieve this by intelligently navigating the energy landscape, concentrating computational effort on regions crucial for understanding the dynamics of these otherwise elusive processes and providing insight into the system’s behavior with limited computational resources.
Conventional approaches to simulating molecular events frequently demand pre-existing knowledge of the system’s behavior, a significant constraint when investigating truly complex phenomena. These methods, while effective in simplified scenarios, often stumble when faced with the intricate energy landscapes characteristic of biological macromolecules or novel chemical reactions. The requirement for initial assumptions – such as identifying likely reaction pathways or key intermediate states – limits the ability to discover unexpected behaviors or explore entirely uncharted territory. Consequently, simulations become heavily biased towards pre-conceived notions, hindering the potential for genuine discovery and necessitating the development of techniques capable of navigating complexity without relying on prior information about the system’s potential states.
Transition Path Sampling: An Unbiased Path to Discovery
Transition Path Sampling (TPS) is a simulation methodology used to explore the pathways between stable states in a dynamic system. Unlike many enhanced sampling techniques, TPS does not require a pre-defined reaction coordinate to identify and sample reactive events. Instead, it directly generates complete trajectories connecting initial and final states by repeatedly simulating short segments, accepting only those that successfully link reactant and product configurations. This approach ensures an unbiased representation of the transition paths, meaning all possible pathways are, in principle, equally accessible during the sampling process, and the statistical weight of each pathway reflects its true probability. The technique relies on Monte Carlo methods to efficiently explore the high-dimensional space of possible trajectories without prior knowledge of the underlying reaction mechanism.
Transition Path Sampling (TPS) generates trajectories representing complete reaction events, allowing for the construction of a network of pathways connecting reactant and product states. Unlike methods focusing on specific coordinates or intermediates, TPS explores configuration space by stringing together consecutive time steps along successful trajectories. This approach inherently captures the diversity of possible pathways, including those that may be rare or involve complex, multi-state transitions. The resulting network is not limited to the most probable path, but rather represents the full ensemble of reactive routes, enabling detailed analysis of reaction mechanisms and the identification of alternative transition states. The connectivity and statistical weight of each pathway within the network directly reflects its contribution to the overall reaction rate.
Transition Path Sampling (TPS), while unbiased, exhibits computational cost that scales significantly with system complexity. The primary driver of this expense is the need to generate and analyze numerous complete trajectories connecting reactant and product states. In high-dimensional configuration spaces, the volume of possible pathways increases exponentially, requiring substantially more sampling to achieve adequate coverage of the transition paths. This is because the probability of finding relevant trajectories decreases as the dimensionality of the system increases, necessitating a greater number of trajectory attempts to locate and characterize the reactive network. Furthermore, the storage and analysis of these trajectories also contribute to the computational burden, particularly when dealing with long or complex pathways.
Waste-Recycling Transition Path Sampling (TPS) enhances computational efficiency by incorporating information from trajectories that do not directly connect reactants to products. Standard TPS discards non-reactive trajectories, but Waste-Recycling TPS reuses portions of these trajectories, specifically segments between states that have already been sampled. This recycling process effectively expands the accessible configuration space without the computational cost of generating entirely new trajectories, thereby increasing the sampling rate of rare reactive events and reducing the overall computational demand for complex systems.
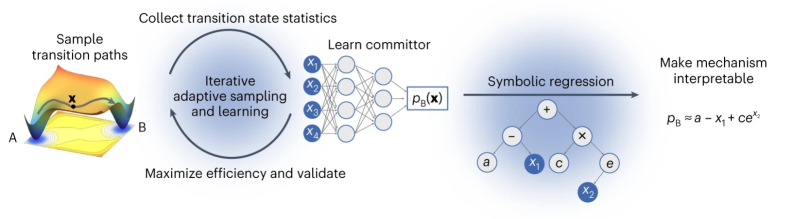
AIMMD: Guiding Discovery with Machine Learning
Artificial Intelligence for Molecular Mechanism Discovery (AIMMD) integrates Transition Path Sampling (TPS) with machine learning techniques to dynamically determine the reaction coordinate during molecular dynamics simulations. Traditional TPS requires a pre-defined collective variable to efficiently sample rare events; AIMMD circumvents this limitation by employing a neural network to learn the optimal reaction coordinate directly from trajectory data. This allows for the identification of relevant pathways without prior knowledge of the system’s behavior. The machine learning component predicts the committor probability – the probability a trajectory will transition from an initial to a final state – and iteratively refines this prediction based on observed trajectories, effectively learning the minimum reaction coordinate on-the-fly and accelerating the sampling process.
The committor probability, [latex]P_{commit}[/latex], quantifies the probability that a molecular trajectory, initiated from a specific state, will reach a target state (B) before returning to its initial state (A). Within the AIMMD framework, a neural network is trained to approximate this value based on the current configuration of the system. The network receives as input a system’s state – defined by atomic coordinates – and outputs a prediction for [latex]P_{commit}[/latex]. This allows for on-the-fly identification of critical regions along the reaction coordinate without requiring prior knowledge of the system’s potential energy surface. The predicted committor probability serves as a collective variable for enhanced sampling, guiding the exploration of relevant conformational space and accelerating the calculation of reaction rates.
The neural network employed in AIMMD is trained using a Negative Log-Likelihood (NLL) loss function to optimize its committor probability predictions. This function quantifies the difference between the predicted committor probabilities and the observed outcomes of trajectories – specifically, whether the trajectory successfully transitioned from state A to state B. Minimizing the NLL effectively encourages the network to assign high probabilities to trajectories that did transition and low probabilities to those that did not. The loss is calculated as [latex] – \sum_{i} [ y_{i} \log(p_{i}) + (1 – y_{i}) \log(1 – p_{i}) ] [/latex], where [latex] y_{i} [/latex] represents the observed outcome (0 or 1) for trajectory i and [latex] p_{i} [/latex] is the network’s predicted committor probability for that trajectory. This iterative refinement, guided by observed trajectories, allows the network to progressively improve its accuracy in predicting transition probabilities.
AIMMD’s transferability was demonstrated through few-shot learning applied to ion association and dissociation processes. The methodology successfully adapted to different monovalent salts with minimal retraining; specifically, only the final layer of the neural network required adjustment for each new salt. This limited retraining significantly reduces computational cost and allows for rapid application of the learned reaction coordinate to related systems, indicating the network effectively generalizes underlying principles governing the transition pathways rather than memorizing specific trajectories.
Transition Path Sampling (TPS) relies on shooting trajectories from intermediate states to map the reaction pathway between two stable states. The selection of these ‘shooting points’ is critical for efficient sampling; AIMMD employs a Lorentzian distribution to govern this process. This distribution provides a balance between exploration – ensuring diverse sampling of conformational space – and exploitation – focusing on regions with high probability of connecting the two states. Specifically, the Lorentzian function’s characteristic long tails facilitate infrequent but crucial sampling of high-energy, rare events, while its peaked central region prioritizes trajectories likely to contribute to the pathway. This approach enhances convergence by effectively navigating the energy landscape and generating a statistically robust representation of the reaction mechanism, compared to uniform or Gaussian sampling methods.
From Committor to Mechanism: Illuminating the Pathways
Accelerated Importance-weighted Markov diffusion (AIMMD) enables a powerful analytical approach to understanding complex reaction mechanisms by leveraging symbolic regression. This technique doesn’t merely identify that a reaction occurs, but elucidates how it happens, by expressing the learned committor – the probability a system will transition to a specific state – as a concise mathematical function of preselected collective variables. Essentially, AIMMD distills the complex dynamics of a system into an interpretable equation, allowing researchers to pinpoint the key degrees of freedom governing the reaction and gain valuable insights into the underlying physical processes. This analytical description moves beyond purely numerical observations, providing a level of mechanistic understanding difficult to achieve with traditional simulation methods and offering a means to predict behavior under varying conditions.
A key benefit of analyzing a reaction through its committor landscape lies in the ability to distill complex dynamics into an interpretable mechanism. Rather than simply observing a transformation, this approach identifies the essential collective variables – the specific coordinates that govern the process – and expresses the probability of transitioning to a product state as a function of those variables. This concise representation, often achieved through techniques like Symbolic Regression, reveals the underlying physical processes driving the reaction, offering insights into rate-limiting steps and key intermediate states. The resulting model isn’t merely descriptive; it provides a predictive framework for understanding how subtle changes in the system might influence the reaction’s trajectory, ultimately accelerating the design of more efficient processes or the stabilization of desired products.
Recent investigations into the assembly of Mga2 proteins leveraged Accelerated Importance Molecular Dynamics (AIMMD) in conjunction with parallel Transition Path Sampling (TPS) simulations to reveal a surprisingly complex reaction landscape. The study demonstrated that Mga2 assembly doesn’t proceed via a single, dominant pathway, but rather through two distinct, competitive routes. AIMMD effectively analyzed the learned committor – a measure of a molecule’s propensity to reach a specific state – allowing researchers to discern these alternate mechanisms. By identifying the collective variables governing the transition between states, the simulations provided detailed insights into the molecular choreography of assembly, showcasing how seemingly minor variations in initial conditions can lead to dramatically different outcomes and highlighting the importance of considering multiple reaction pathways in understanding complex biological processes.
The successful application of advanced sampling methods, such as those employed in analyzing complex molecular dynamics, relies heavily on specialized software infrastructure. Packages like OpenPathSampling and PyRETIS provide the tools necessary to implement and rigorously analyze these techniques, effectively bridging the gap between theoretical frameworks and practical computation. OpenPathSampling streamlines the process of setting up and executing path sampling simulations, while PyRETIS focuses on the efficient calculation and analysis of rare event properties, including reaction rates and free energy landscapes. These software solutions not only automate complex procedures but also offer functionalities for data analysis, visualization, and validation, empowering researchers to extract meaningful insights from computationally demanding simulations and accelerate discoveries in fields like chemistry, biology, and materials science.
Establishing the reliability of a learned reaction coordinate requires careful validation, typically achieved by comparing the committor values predicted by the analytical model with those numerically estimated from extensive simulations. While strong alignment between predicted and estimated values bolsters confidence in the coordinate’s accuracy, it’s crucial to recognize this process isn’t foolproof; a good fit doesn’t definitively prove the coordinate perfectly captures the underlying reaction mechanism. Discrepancies can arise from limitations in the simulation data, the complexity of the free energy landscape, or the inherent approximations within the analytical method itself. Therefore, this validation serves as a strong indicator, but should be considered alongside other analytical insights and physical intuition when interpreting the results.
The pursuit of efficiently simulating rare molecular events, as detailed in this work, echoes a fundamental principle of scientific inquiry. James Clerk Maxwell observed, “The true value of any theory is not in its ability to predict, but in its ability to explain.” AIMMD, by integrating machine learning with transition path sampling, doesn’t merely accelerate computation; it attempts to explain the mechanisms governing these rare events through the automated discovery of reaction coordinates. This aligns with a view that technological advancement must be paired with interpretability, ensuring that the ‘acceleration’ offered by AI is guided by a deeper understanding of the underlying phenomena, rather than simply a faster path to an unknown outcome. The method’s ability to discern these coordinates reflects a commitment to illuminating, not just calculating.
Beyond the Bottleneck
The automation of reaction coordinate discovery, as demonstrated by AIMMD, represents a tactical victory, yet obscures a more fundamental challenge. The field frequently fixates on efficiently simulating rare events, often neglecting to rigorously examine which events deserve such focused computational resources. Every algorithmic choice-the specific machine learning model, the committor threshold, even the definition of ‘rare’-implicitly encodes a prioritization of molecular behavior. This work, while demonstrably effective, does not alleviate the ethical responsibility to define worthwhile simulations, or to consider the potential consequences of accelerating certain molecular pathways over others.
Future development will undoubtedly focus on extending AIMMD’s capabilities-handling increasingly complex systems, incorporating more diverse data types, and improving the robustness of the learned reaction coordinates. However, a truly conscious progression requires parallel effort in meta-analysis. The field needs standardized methods for assessing the inherent biases within these algorithms, and for quantifying the uncertainty associated with automatically discovered reaction coordinates. Simply accelerating discovery without understanding its inherent limitations feels… familiar.
The promise of artificial intelligence lies not in replacing scientific intuition, but in augmenting it. AIMMD offers a powerful tool, but its ultimate value will be determined by the foresight applied to its use. The next step isn’t merely faster simulations, but a more deliberate conversation about the molecular stories the field chooses to tell.
Original article: https://arxiv.org/pdf/2602.05167.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- The Division Resurgence Best Weapon Guide: Tier List, Gear Breakdown, and Farming Guide
- Last Furry: Survival redeem codes and how to use them (April 2026)
- Clash of Clans Sound of Clash Event for April 2026: Details, How to Progress, Rewards and more
- Guild of Monster Girls redeem codes and how to use them (April 2026)
- GearPaw Defenders redeem codes and how to use them (April 2026)
- Kagurabachi Chapter 118 Release Date, Time & Where to Read Manga
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- Gold Rate Forecast
- eFootball 2026 “Countdown to 1 Billion Downloads” Campaign arrives with new Epics and player packs
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
2026-02-06 23:25