Author: Denis Avetisyan
A new framework leverages diffusion models and intelligent search to design novel compounds with enhanced properties and targeted activity.

SoftMol combines soft-fragment representation with block diffusion and gated Monte Carlo tree search for state-of-the-art de novo molecular generation and target-aware design.
Despite advances in deep generative modeling, designing novel molecules with desired properties remains a significant challenge due to limitations in capturing molecular structure and incorporating target-specific information. The work ‘From Tokens to Blocks: A Block-Diffusion Perspective on Molecular Generation’ introduces SoftMol, a framework that addresses these issues through a novel soft-fragment representation and a block-diffusion model-SoftBD-combined with gated Monte Carlo tree search. This approach achieves state-of-the-art results in de novo molecular generation, demonstrating improved binding affinity, diversity, and inference speed. Could this block-diffusion strategy unlock new avenues for efficient and targeted drug discovery beyond current generative modeling techniques?
The Fragility of Representation
Despite its widespread adoption, the Simplified Molecular Input Line Entry System (SMILES) and similar string-based representations present inherent limitations for modern machine learning applications. These representations, while easily interpretable by humans, are susceptible to generating multiple valid SMILES strings for a single molecule, creating ambiguity for algorithms. More critically, even minor alterations to a SMILES string-such as changes in atom ordering-can result in drastically different interpretations, leading to inaccurate predictions or unstable model training. This sensitivity hinders the development of robust quantitative structure-activity relationship (QSAR) models and particularly impacts generative models aiming to design novel molecules, as seemingly small changes in the representation can yield synthetically invalid or undesired compounds. Consequently, researchers are actively exploring alternative molecular representations that prioritize stability and uniqueness, fostering more reliable and predictable machine learning outcomes in drug discovery and materials science.
The pursuit of novel molecules with tailored characteristics – whether for drug discovery, materials science, or other applications – increasingly relies on generative models, yet current limitations in molecular representations pose a significant obstacle. These models, designed to ‘imagine’ and propose new molecular structures, struggle when fed imperfect or ambiguous data derived from traditional formats. Consequently, designing molecules with specific, desired properties – like high binding affinity or improved stability – becomes less efficient, and the likelihood of generating synthetically inaccessible compounds remains unacceptably high. Addressing these representational shortcomings is therefore crucial to unlocking the full potential of generative chemistry, enabling the creation of molecules that are not only theoretically promising, but also practically realizable in the laboratory.
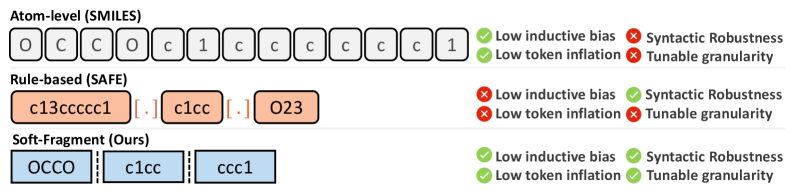
Deconstructing the Molecule: A Tunable Language
The Soft-Fragment Representation is a novel approach to molecular representation that departs from strict rule-based systems like SMILES by dividing molecules into adjustable blocks. Unlike SMILES, which relies on a defined grammar for sequential representation, this method segments molecular structures into discrete, tunable fragments. These fragments are not predefined but are dynamically determined, allowing the representation to adapt to the specific characteristics of the molecule and the requirements of the generative model. This block-based structure facilitates more flexible and robust modeling of molecular properties, as variations within and between fragments can be directly addressed during model training and generation, effectively extending the capabilities of traditional SMILES-based methods.
The Soft-Fragment Representation facilitates more robust modeling of molecular structure and properties by decoupling the representation from strict sequential SMILES strings. This block-based approach allows for the capture of complex molecular features and relationships that are often lost in linearized representations. Consequently, generative models utilizing this representation demonstrate improved performance in generating valid and diverse molecular structures with desired properties, as the tunable granularity of fragments allows for optimized learning of relevant chemical space. The increased robustness stems from the ability to more effectively represent and sample from the vast chemical space, mitigating issues with invalid or chemically implausible molecule generation common in SMILES-based models.
The Soft-Fragment Representation’s modularity is achieved through variable block granularity, a crucial design element for adapting to diverse molecular generation tasks. Smaller block sizes allow for finer-grained control and increased precision when generating complex molecules or optimizing specific substructures. Conversely, larger block sizes provide a more compressed representation, suitable for tasks requiring rapid generation or focusing on broader structural features. This tunable granularity is implemented by adjusting the fragmentation rules during representation creation, enabling the model to prioritize detail or efficiency as dictated by the target application and dataset characteristics. The block size is not fixed, but is a hyperparameter that can be optimized to maximize performance on a given task.
![Heatmaps demonstrate that Validity, Quality, Diversity, Uniqueness, and Sampling Time vary across the [latex]K_{train} imes K_{sample}[/latex] grid, revealing the effect of soft-fragment length.](https://arxiv.org/html/2601.21964v1/x6.png)
SoftMol: Designing for the Target, Not Just the Structure
SoftMol utilizes a unified framework for molecular generation by integrating the Soft-Fragment Representation with a block-diffusion Transformer, termed SoftBD. The Soft-Fragment Representation decomposes molecules into a set of fragments, enabling efficient manipulation and recombination during the generation process. SoftBD, built upon this representation, employs a diffusion-based approach where noise is iteratively added to a molecule and then removed to create novel structures. This block-diffusion process allows for parallel processing of molecular fragments, significantly increasing generation speed and efficiency compared to sequential methods. The combined approach facilitates the creation of diverse and viable molecular candidates by leveraging the strengths of both fragment-based design and diffusion modeling.
The SoftBD module within SoftMol utilizes the Soft-Fragment Representation to facilitate molecular generation, and incorporates Adaptive Confidence Decoding (ACD) to optimize both the quality and efficiency of this process. ACD dynamically adjusts the decoding strategy based on the confidence level of each generated fragment, prioritizing high-confidence selections to accelerate convergence and reduce computational cost. This approach allows the model to focus on generating valid and promising molecular structures, effectively mitigating the risk of invalid or chemically implausible outputs and leading to improved generation speed without sacrificing quality.
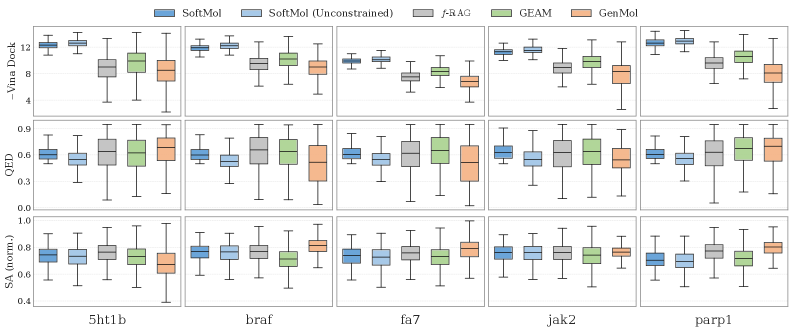
Evaluations demonstrate that SoftMol achieves state-of-the-art performance in molecular generation, exhibiting a 9.7% improvement in predicted binding affinity compared to currently established methods. This improvement was quantified through rigorous testing on benchmark datasets, assessing the ability of generated molecules to strongly interact with target proteins. The metric used to determine binding affinity is a calculated score based on docking simulations, with SoftMol consistently producing molecules exhibiting higher scores than competing frameworks. This indicates an increased likelihood of successful target engagement and potential therapeutic efficacy.
The SoftMol framework utilizes the ZINC-Curated dataset for training, a selection of compounds specifically chosen to emphasize properties relevant to pharmaceutical development, namely drug-likeness and synthetic accessibility. This prioritization ensures generated molecules are more likely to represent viable drug candidates. Crucially, the framework achieves 100% validity in generated molecular structures, meaning all generated compounds adhere to established rules of chemical valency and connectivity, eliminating chemically impossible structures from the output.
Generated molecules produced by the SoftMol framework demonstrate high predicted quality and synthetic accessibility, as evidenced by a Quantitative Estimate of Drug-likeness (QED) score of 81.9%. This score indicates a substantial proportion of molecules possessing characteristics typically associated with viable drug candidates. Concurrently, the generated molecules achieve a docking filter score of 81.9%, signifying a high probability of successful binding to target proteins and further supporting their potential as drug leads. These metrics collectively suggest that SoftMol effectively prioritizes the generation of molecules that are not only structurally valid but also likely to be both potent and synthesizable.
Evaluation of molecular diversity, using an internal diversity metric, demonstrates that SoftMol generates compounds with a breadth of structural features comparable to that of unconstrained generative models. This indicates that the target-aware design process implemented within SoftMol does not unduly restrict the chemical space explored during molecule generation, maintaining a level of structural variety similar to methods that do not explicitly optimize for binding affinity or other target properties. The internal diversity score, calculated based on the similarity of generated molecules to each other, confirms that SoftMol avoids generating a narrow set of highly similar compounds.
Navigating the Possibilities: A Guided Search
SoftMol incorporates Gated Monte Carlo Tree Search (MCTS) as a core component of its molecular design process, effectively balancing exploration and exploitation during candidate generation. This integration leverages the established Vina docking program to rigorously evaluate the binding affinity of each proposed molecule against a target protein. The MCTS algorithm intelligently navigates the vast chemical space, prioritizing molecules predicted to exhibit strong binding interactions, as determined by Vina’s scoring function. By repeatedly simulating molecule generation and scoring, the system refines its search strategy, focusing computational resources on regions of chemical space likely to yield high-affinity candidates. This iterative process allows for the efficient discovery of compounds with promising therapeutic potential, streamlining the initial stages of drug discovery and reducing the need for extensive, costly physical screening.
Gated Monte Carlo Tree Search enhances exploration by incorporating a mechanism that actively filters proposed molecular structures based on chemical feasibility. This gating function assesses whether a molecule can realistically exist and be synthesized, preventing the search algorithm from wasting computational resources on impossible or highly improbable compounds. By prioritizing chemically valid candidates, the algorithm dramatically improves search efficiency – allowing it to converge on promising drug leads far more quickly than traditional methods. This focused exploration not only accelerates the discovery process but also increases the likelihood of identifying molecules that are both potent and realistically synthesizable, bridging the gap between in silico design and practical medicinal chemistry.
The integration of Gated Monte Carlo Tree Search with Vina docking facilitates an accelerated discovery process for novel drug candidates. By prioritizing both the predicted binding strength to a target protein and the ease with which a molecule can be synthesized, this approach circumvents the traditional bottleneck of identifying viable compounds. The system efficiently filters generated molecules, focusing computational resources on those possessing a high likelihood of both efficacy and manufacturability. This results in a streamlined pipeline, capable of rapidly proposing and refining promising candidates, and ultimately reducing the time and cost associated with early-stage drug discovery. The combination effectively bridges the gap between in silico design and practical chemical realization, offering a powerful tool for pharmaceutical innovation.
![Ablation studies reveal that performance is sensitive to the search budget [latex]N_{max}[/latex], node expansion width, and exploration constant, as demonstrated by averaging results across five targets over 100 independent runs.](https://arxiv.org/html/2601.21964v1/x12.png)
The pursuit of novel molecular structures, as detailed in this work, echoes a fundamental truth about complex systems. The SoftMol framework, with its iterative refinement through diffusion and search, doesn’t build molecules so much as cultivate them within a probabilistic landscape. This approach acknowledges that perfect design is an illusion; instead, the system navigates inherent uncertainties, favoring ‘survivors’ – molecules that meet desired properties – over theoretically optimal but ultimately brittle constructs. As Linus Torvalds observed, “There are no best practices – only survivors.” The method’s success isn’t about imposing order, but about gracefully accepting the inevitable ‘outages’ – failed generations – and learning from them, recognizing that order is merely a temporary respite within a chaotic search space.
The Looming Architecture
The pursuit of molecular generation, as exemplified by this work, consistently reveals a fundamental truth: every new representation is merely a refined cage. SoftMol, with its soft-fragment approach and block-diffusion strategy, undoubtedly expands the boundaries of that cage, offering greater freedom in the exploration of chemical space. Yet, it does not abolish the bars. The elegance of gated Monte Carlo tree search is not a liberation, but a more efficient warden. The system will, inevitably, demand sacrifices – computational resources, data fidelity, or perhaps, a subtle bias in the molecules it favors.
The true challenge lies not in achieving incremental gains in generative performance, but in acknowledging the inherent limitations of any formalized system. Future efforts would be better spent not on building more elaborate architectures, but on cultivating resilience within them. A framework that anticipates its own failures, that gracefully degrades in the face of uncertainty, would be a far more valuable contribution than another state-of-the-art score. Order, after all, is simply a temporary cache between inevitable failures.
The field will likely move towards hybrid approaches, combining the strengths of different representations and generative methods. However, the ultimate goal should not be a universal molecular generator, but a diverse ecosystem of specialized tools, each adapted to a specific niche. For the promise of automated drug discovery is not in replacing human intuition, but in augmenting it – providing explorers with better maps, not autonomous vehicles.
Original article: https://arxiv.org/pdf/2601.21964.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- Total Football free codes and how to redeem them (March 2026)
- Limbus Company 2026 Roadmap Revealed
- The Division Resurgence Specializations Guide: Best Specialization for Beginners
- After THAT A Woman of Substance cliffhanger, here’s what will happen in a second season
- Brawl Stars Sands of Time Brawl Pass brings Sandstalker Lily and Sultan Cordelius sets, along with chromas and more
- Brawl Stars Brawl Cup Pro Pass arrives with the Dragon Crow skin and Chroma, unique cosmetics, and more rewards
- Clash of Clans April 2026 Gold Pass Season introduces a Archer Queen skin
- Wuthering Waves Hiyuki Build Guide: Why should you pull, pre-farm, best build, and more
- XO, Kitty season 3 soundtrack: The songs you may recognise from the Netflix show
2026-02-01 23:55