Author: Denis Avetisyan
Researchers have created an AI system capable of independently discovering the principles governing the design of organic photocatalysts for efficient hydrogen production.
An agentic AI, integrating large language models and quantum chemistry, autonomously derives interpretable structure-property relationships for organic photocatalyst design.
Despite the vastness of chemical space, rational design of high-performance organic photocatalysts remains a significant challenge. This work introduces ‘ChemNavigator: Agentic AI Discovery of Design Rules for Organic Photocatalysts’, an agentic AI system that autonomously uncovers structure-property relationships governing photocatalytic activity through hypothesis-driven exploration and quantum mechanical calculations. ChemNavigator successfully identified six statistically significant design rules-relating to functional groups and electronic structure-without explicit programming, validating principles like resonance and inductive effects. Could this framework, mirroring the scientific method, accelerate materials discovery by complementing-rather than replacing-chemical intuition and enabling the prioritization of synthetic efforts?
Harnessing Light: The Promise of Catalytic Hydrogen
The escalating global demand for energy is driving intense research into sustainable fuel sources, and hydrogen holds significant promise as a clean alternative to fossil fuels. However, current hydrogen production methods often rely on unsustainable processes. Photocatalytic water splitting-the use of light and a catalyst to separate water into hydrogen and oxygen-represents a potentially transformative approach. This method mimics photosynthesis, harnessing solar energy to drive a chemical reaction that yields clean fuel, offering a pathway toward a closed-loop, renewable energy system. While still facing challenges in terms of efficiency and scalability, photocatalytic hydrogen production is gaining momentum as a viable solution to meet future energy needs and mitigate the environmental impact of conventional fuel sources.
Conventional photocatalysts, frequently based on inorganic semiconductors, have historically presented significant obstacles to the widespread production of hydrogen via water splitting. These materials often exhibit limited light absorption capabilities, meaning a substantial portion of solar energy goes unused, and their performance is heavily influenced by factors like particle size and surface defects – characteristics difficult to precisely control. Furthermore, modifying these materials to enhance their efficiency or tailor their properties for specific applications proves challenging due to their rigid structures and limited compositional flexibility. This lack of tunability restricts the optimization process, preventing researchers from fine-tuning the catalyst to maximize hydrogen evolution rates and overall system performance, ultimately hindering the practical implementation of photocatalytic hydrogen production on a large scale.
The pursuit of sustainable energy sources has led researchers to explore organic photocatalysts as a viable alternative to traditional materials for hydrogen production. Unlike their inorganic counterparts, organic photocatalysts boast remarkable structural diversity, allowing for precise molecular engineering to enhance light absorption and charge separation-critical steps in the water-splitting process. This tunability enables scientists to tailor the catalyst’s properties, such as its redox potential and light-harvesting capabilities, through subtle modifications to its molecular architecture. Consequently, organic photocatalysts present a compelling pathway towards achieving higher efficiencies and overcoming the limitations inherent in conventional materials, paving the way for scalable and cost-effective hydrogen fuel production.
The rational design of effective organic photocatalysts hinges on a nuanced comprehension of how molecular structure dictates performance characteristics. Subtle alterations to a molecule’s architecture – including the arrangement of electron-donating and electron-withdrawing groups, the extent of π-conjugation, and the overall three-dimensional shape – profoundly influence its light absorption capabilities, charge separation efficiency, and catalytic activity. Researchers are increasingly employing computational modeling and systematic synthesis to establish clear structure-property relationships, allowing for the targeted optimization of organic photocatalysts for enhanced hydrogen production. This approach moves beyond trial-and-error methods, enabling the creation of materials with precisely tuned properties that maximize the conversion of sunlight into clean, sustainable energy, and paving the way for scalable photocatalytic water splitting technologies.
Autonomous Design: An Agentic AI Platform
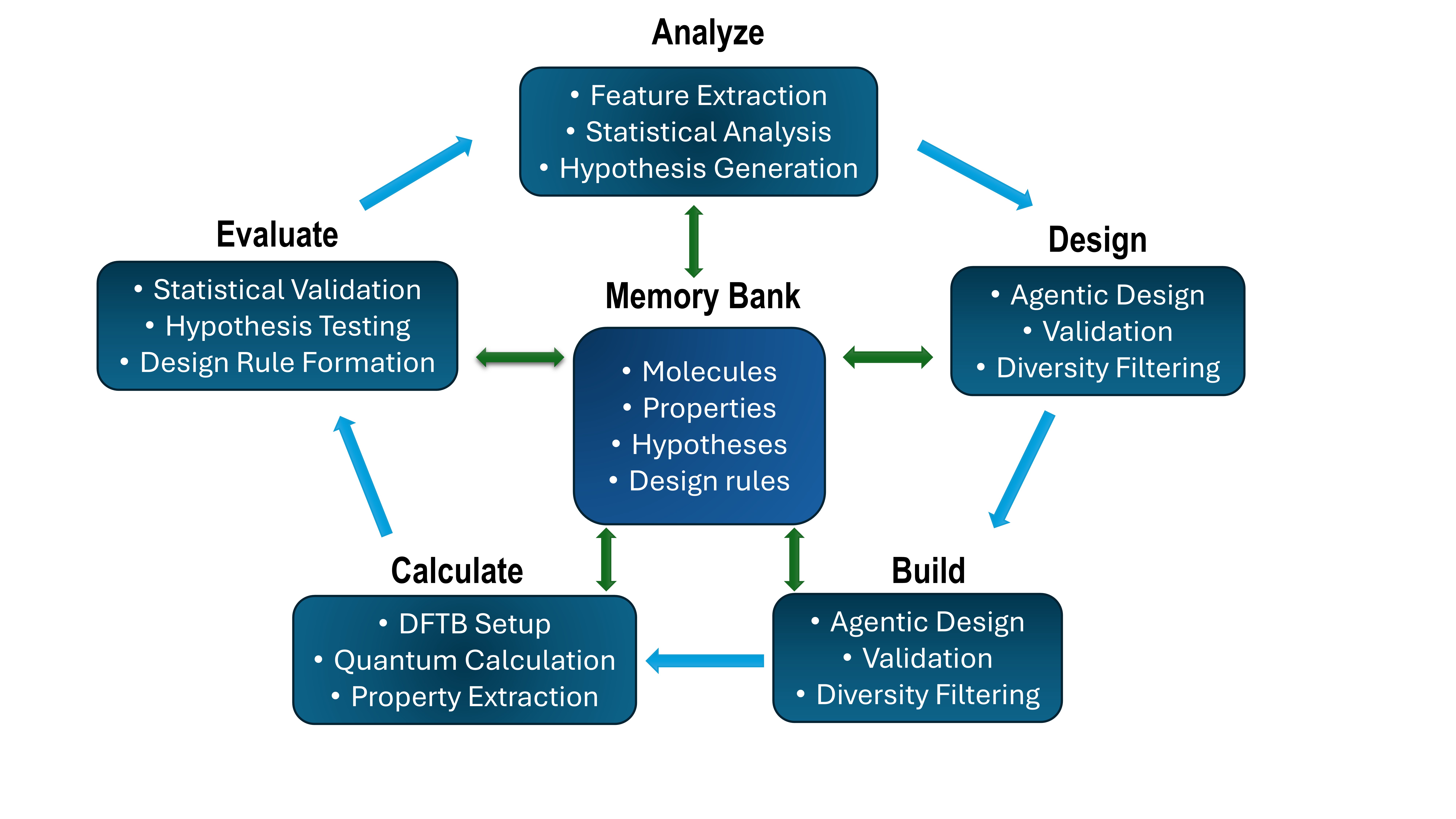
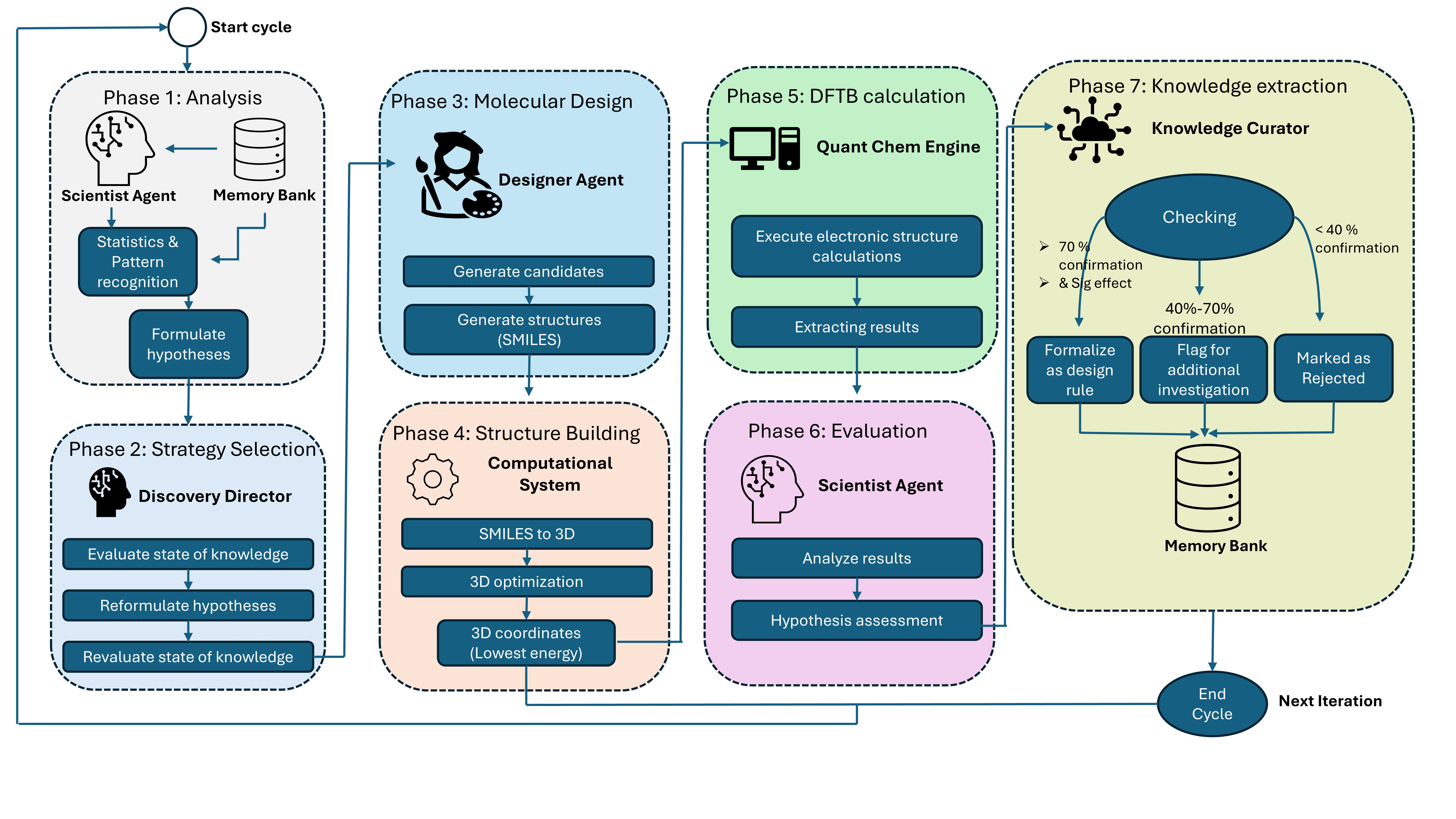
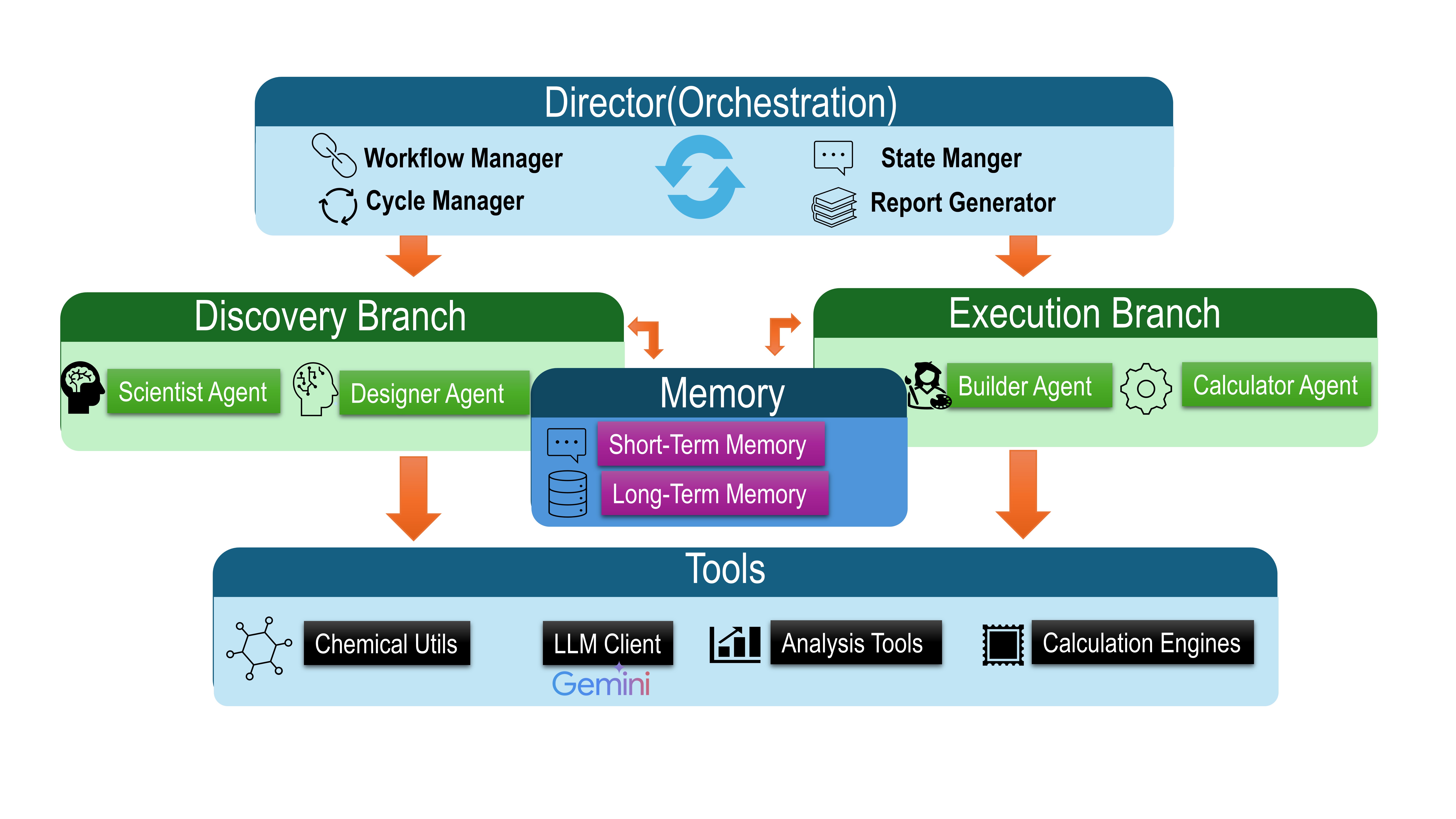
Agentic AI is a new system designed for materials discovery that combines the reasoning capabilities of large language models (LLMs) with the predictive power of density functional theory (DFT) calculations. This integration allows the system to move beyond traditional, human-driven workflows by autonomously formulating and testing hypotheses. The LLM component is used for tasks requiring complex reasoning and knowledge representation, while DFT calculations provide accurate, physics-based predictions of material properties. By coupling these two distinct approaches, Agentic AI facilitates an iterative discovery process where LLM-generated designs are validated and refined through DFT-based assessment, enabling efficient exploration of chemical space.
Agentic AI utilizes a Multi-Agent Architecture comprising four distinct agent types to facilitate autonomous materials discovery. The Hypothesis Agent formulates initial design principles based on desired material properties. The Molecular Design Agent then generates candidate molecular structures adhering to these principles. Subsequently, the Calculation Agent performs density functional theory (DFT) calculations to predict the properties of these designed molecules. Finally, the Evaluation Agent assesses the calculated properties against the initial design criteria, providing feedback to refine the hypothesis and initiate the next iteration of the design-build-test cycle. This modular design allows for parallelization and specialization, improving the efficiency of the overall discovery process.
Agentic AI facilitates the autonomous exploration of chemical space by iteratively generating, calculating, and evaluating potential photocatalyst candidates. The system completed a full discovery cycle – encompassing hypothesis generation, molecular design, DFT calculation, and performance evaluation – for ten distinct molecules within 80 seconds. This throughput is achieved through the coordinated operation of specialized agents within a multi-agent architecture, enabling rapid iteration and screening of chemical possibilities without manual intervention. The automated cycle allows for significantly increased efficiency in materials discovery compared to traditional methods.
Traditional materials discovery is often limited by iterative cycles of human-driven hypothesis, calculation, and analysis, creating significant bottlenecks in the design process. Agentic AI addresses these limitations through automation; its multi-agent system autonomously generates molecular hypotheses, performs density functional theory (DFT) calculations to assess viability, and evaluates results without human intervention. This closed-loop system completed a full discovery cycle for ten molecules in 80 seconds, demonstrating a substantial acceleration compared to conventional methods where each step relies on manual execution and analysis, and thereby reducing the time required to identify potential materials.
Decoding Efficiency: Frontier Orbital Energies and Band Gaps
The efficiency of organic photocatalysts is directly determined by their Frontier Orbital Energies – specifically the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO). These energies define the catalyst’s redox potentials, which govern its ability to accept or donate electrons during photocatalytic reactions. The HOMO energy represents the tendency to donate electrons – its position dictates the strength of the reducing agent. Conversely, the LUMO energy indicates the propensity to accept electrons, thus defining the strength of the oxidizing agent. A smaller band gap – the energy difference between the HOMO and LUMO – facilitates easier excitation of electrons by photons of lower energy, increasing light absorption and photocatalytic activity. Consequently, manipulating these frontier orbital energies is crucial for designing highly efficient organic photocatalysts.
Effective photocatalytic performance relies on optimizing the absorption of light and the subsequent separation of photo-generated charge carriers. Modulating the energies of the Frontier Molecular Orbitals – specifically the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) – directly impacts these processes. A smaller band gap, defined as the energy difference between the HOMO and LUMO, generally allows for absorption of lower energy photons, increasing light harvesting efficiency. However, excessively small band gaps can lead to rapid electron-hole recombination, reducing charge separation efficiency. Conversely, larger band gaps require higher energy photons but can promote more efficient charge separation. Therefore, careful tuning of the HOMO and LUMO energies, and consequently the band gap, is essential to balance light absorption and charge separation, maximizing overall photocatalytic activity.
The system employs Density Functional Theory (DFT) calculations to predict Frontier Molecular Orbital (FMO) energies and band gaps of organic photocatalysts. These calculations are rigorously validated against the established B3LYP functional with the def2-SVP basis set. Quantitative performance metrics demonstrate a strong correlation between the AI-predicted values and the DFT calculations, with Pearson correlation coefficients of 0.9410 for Highest Occupied Molecular Orbital (HOMO) energies, 0.9934 for Lowest Unoccupied Molecular Orbital (LUMO) energies, and 0.9274 for band gap estimations. This high degree of correlation confirms the reliability and predictive power of the Agentic AI system for materials discovery and optimization.
Analysis reveals a direct correlation between the inclusion of specific functional groups and alterations in the Frontier Orbital energies of organic photocatalysts. Incorporation of Carbonyl groups consistently lowers the Highest Occupied Molecular Orbital (HOMO) energy, increasing the material’s oxidative power. Conversely, the introduction of Ether groups primarily impacts the Lowest Unoccupied Molecular Orbital (LUMO) energy, influencing reductive capabilities and consequently affecting the Band Gap – the energy difference between the HOMO and LUMO. These modifications, achieved through systematic functionalization, provide a pathway to tune the optoelectronic properties of the photocatalyst for enhanced performance in specific applications.
![Combining electron-donating ether linkages and electron-withdrawing carbonyl groups results in a synergistic reduction of the band gap ([latex]2.66 ext{ eV}[/latex]) that surpasses the expected additive effect ([latex]1.97 ext{ eV}[/latex]), suggesting diminishing returns from combined functionalization.](https://arxiv.org/html/2601.17084v1/interaction_plot_final_fix.jpg)
From Molecules to Principles: Deriving Design Rules
Agentic AI has moved beyond traditional trial-and-error methods in photocatalyst design by systematically exploring the vast chemical space of organic molecules. This approach yielded a set of statistically validated Design Rules that govern performance, offering predictive power previously unattainable. Instead of relying on expert intuition, the AI identified correlations between molecular features and photocatalytic activity, uncovering principles like the impact of ether linkages on [latex]HOMO[/latex] energies and carbonyl groups on band gaps. The resulting rules, established with large effect sizes – exceeding 0.8 and, in some cases, reaching 1.42 – provide a framework for rationally designing more efficient organic photocatalysts, accelerating discovery and potentially revolutionizing fields like solar energy conversion and pollution remediation.
The systematic investigation into organic photocatalyst design consistently reveals extended conjugation as a powerful strategy for optimizing light absorption and, consequently, performance. This principle centers on the creation of delocalized π-electron systems within the molecular structure; by increasing the number of atoms participating in this electron cloud, the energy required for electron excitation – the Band Gap – is predictably lowered. A smaller Band Gap allows the material to absorb lower-energy photons from the visible light spectrum, dramatically increasing its efficiency in photocatalytic reactions. The data indicates a strong correlation between extended conjugation and improved light harvesting, suggesting this design rule is fundamental to achieving high-performing organic photocatalysts and representing a crucial step towards sustainable energy solutions.
Halogenation, the incorporation of fluorine, chlorine, bromine, or iodine into a molecular structure, presents a nuanced pathway for manipulating the performance of organic photocatalysts. While seemingly straightforward, the effects of halogenation are far from simple, often involving a complex interplay of inductive and resonance effects that influence [latex]Frontier\,Orbital\,Energies[/latex]. Strategic halogenation can effectively lower the energy of the Lowest Unoccupied Molecular Orbital (LUMO) and/or raise the energy of the Highest Occupied Molecular Orbital (HOMO), thereby narrowing the band gap and enhancing light absorption. This fine-tuning capability is critical, as even subtle shifts in these energy levels can dramatically impact a photocatalyst’s redox potential and overall efficiency. Agentic AI’s systematic investigations reveal that careful selection of halogen type and placement allows for predictable and substantial control over these crucial electronic properties, optimizing photocatalytic activity without necessarily increasing molecular complexity.
Predictive accuracy in organic photocatalyst design hinges on realistically simulating the solvent environment, a challenge effectively addressed through the GBSA Implicit Solvation Model. Recent investigations leveraging this model have yielded six statistically validated design rules – all with p-values below 0.05 and substantial effect sizes (d ≥ 0.8) – revealing critical molecular features impacting performance. Notably, the presence of ether linkages demonstrates a particularly strong influence, shifting Highest Occupied Molecular Orbital (HOMO) energies by 1.42, while carbonyl groups significantly reduce Band Gaps – as quantified by an effect size of -1.13. These findings underscore the importance of accounting for solvation when translating molecular structure into functional photocatalytic materials, offering a robust framework for targeted design and performance optimization.
A Future of Autonomous Materials Innovation
The conventional process of materials discovery has long relied on exhaustive experimentation – a costly and time-consuming cycle of synthesis, characterization, and analysis. Agentic AI introduces a fundamental shift, transitioning this field from a predominantly trial-and-error approach to one grounded in rational design principles. Instead of randomly testing material combinations, this innovative system autonomously navigates the vast landscape of chemical possibilities, guided by its own evolving understanding of structure-property relationships. It doesn’t simply analyze existing data; it formulates hypotheses, designs experiments to test those hypotheses, and iteratively refines its knowledge – effectively acting as an autonomous scientist. This capability allows for the targeted creation of materials with pre-defined characteristics, dramatically accelerating the pace of innovation and potentially unlocking solutions to previously intractable challenges.
The advent of systems capable of independently navigating the vast landscape of chemical possibilities represents a pivotal advancement in materials science. Rather than relying on iterative experimentation or human-guided searches, this technology autonomously proposes, evaluates, and refines material compositions, effectively charting a course through chemical space. This process isn’t merely about discovering new materials; it’s about deriving the fundamental principles that govern material behavior. By identifying correlations between chemical structure and desired properties, the system establishes design rules that transcend individual discoveries, allowing for the rational creation of materials tailored to specific applications. This capability unlocks unprecedented opportunities for innovation, promising to dramatically accelerate the development of advanced technologies and address complex challenges across numerous scientific disciplines.
The potential of Agentic AI extends far beyond the initial demonstration in magnetic materials, offering a pathway to dramatically accelerate innovation across a spectrum of functional materials. Researchers anticipate applying this autonomous design framework to challenges in energy storage, where optimizing materials for batteries and supercapacitors demands navigating complex chemical spaces. Similarly, in the biomedical field, Agentic AI could revolutionize the development of novel drug delivery systems, biocompatible implants, and advanced tissue engineering scaffolds. By intelligently exploring material compositions and structures, the system bypasses conventional limitations and promises to unlock materials with tailored properties for applications ranging from efficient solar cells to targeted therapies, fundamentally reshaping how materials are discovered and deployed to address global challenges.
The convergence of artificial intelligence and scientific computation represents a powerful new approach to tackling complex global challenges. This research demonstrates how agentic AI, operating within the framework of computational materials science, can move beyond simply analyzing existing data to actively designing novel materials with desired properties. By autonomously formulating hypotheses, conducting virtual experiments, and interpreting results, the system accelerates the innovation process – a feat previously limited by the constraints of human intuition and experimental throughput. This integration isn’t merely about speed; it unlocks access to previously unexplored regions of chemical space, potentially yielding breakthroughs in areas like sustainable energy, advanced medicine, and environmental remediation. The capacity to computationally predict material behavior, coupled with AI’s ability to navigate complex datasets and optimize designs, offers a scalable and efficient pathway toward solving critical issues facing humanity.
The pursuit of efficient photocatalysts, as detailed in this research, benefits from a relentless simplification of complex data. The agentic AI presented doesn’t merely find design rules; it distills them from quantum chemical calculations, revealing underlying principles. This echoes Richard Feynman’s sentiment: “The first principle is that you must not fool yourself – and you are the easiest person to fool.” The AI, by grounding its discoveries in fundamental calculations, avoids the pitfalls of purely empirical observation. It seeks an understanding, not just a correlation, offering interpretable design rules – a clarity achieved through the removal of unnecessary complexity, much like striving for elegance in both scientific discovery and computational design. The focus on structure-property relationships exemplifies this desire for fundamental understanding.
The Road Ahead
The demonstration of autonomous discovery, while encouraging, merely clarifies the scale of what remains unknown. The current work establishes a functional, if limited, bridge between language and calculation. The true challenge lies not in generating more photocatalyst designs, but in refining the criteria for meaningful design. The system presently validates rules against a single metric-hydrogen evolution efficiency. A more stringent test will demand generalization across diverse catalytic reactions and operating conditions, forcing a deeper understanding of transferable design principles.
Further progress requires confronting the inherent limitations of the underlying approximations. Density functional tight binding, while computationally efficient, introduces errors. The system’s “discoveries” are, therefore, provisional-rules validated within a specific level of theory. The pursuit of more accurate, yet still scalable, quantum chemical methods is paramount. Equally important is the development of methods to quantify uncertainty-to know not just what the system discovers, but how confident it is in those discoveries.
Ultimately, the field must move beyond the optimization of individual molecules. The design of functional materials demands consideration of macroscopic properties-stability, processability, and cost. Integrating these considerations into an agentic AI framework will necessitate a more holistic representation of materials-one that transcends the limitations of simple structure-property relationships. The goal is not intelligent design, but rather, the intelligent reduction of complexity.
Original article: https://arxiv.org/pdf/2601.17084.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Beyond Accuracy: Gauging Trust in Human-AI Teams
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- How Martin Clunes has been supported by TV power player wife Philippa Braithwaite and their anti-nepo baby daughter after escaping a ‘rotten marriage’
- Gold Rate Forecast
- CookieRun: OvenSmash coupon codes and how to use them (March 2026)
- Clash Royale Balance Changes March 2026 — All Buffs, Nerfs & Reworks
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- Total Football free codes and how to redeem them (March 2026)
- Only One Straw Hat Hasn’t Been Introduced In Netflix’s Live-Action One Piece
- Genshin Impact Version 6.5 Leaks: List of Upcoming banners, Maps, Endgame updates and more
2026-01-27 09:21