Author: Denis Avetisyan
A new approach combines artificial intelligence with strategic tool use to significantly advance the design and synthesis of complex molecules.

This research introduces ChemCRAFT, a framework leveraging agentic reinforcement learning to decouple chemical reasoning from computation and optimize tool usage for expert-level molecular problem-solving.
Despite recent advances, current chemical language models struggle with a trade-off between knowledge retention and computational cost, hindering their application in complex molecular design and synthesis tasks. This work introduces ChemCRAFT, a novel framework detailed in ‘Agentic reinforcement learning empowers next-generation chemical language models for molecular design and synthesis’, which leverages agentic reinforcement learning to decouple chemical reasoning from knowledge storage, enabling locally deployable small models to achieve expert-level performance. By training a language model to strategically utilize external tools within a chemical sandbox-guided by a novel [latex]SMILES-GRPO[/latex] reward function-we demonstrate superior results in molecular structure analysis, optimization, and synthesis prediction compared to existing cloud-based large language models. Could this approach to tool-augmented reasoning unlock a new paradigm for cost-effective and privacy-preserving AI-aided chemistry, accelerating molecular discovery beyond the limitations of model scale?
The Limits of Pattern Recognition: Why Scaling Isn’t Enough
Large language models have demonstrated a remarkable capacity for identifying patterns within vast datasets, a skill that initially suggested promise for complex scientific domains like chemistry. However, true chemical reasoning extends far beyond mere pattern recognition; it requires an understanding of underlying principles, the ability to extrapolate from limited data, and the capacity to navigate a space of possibilities governed by physical laws. Unlike tasks where memorization suffices, chemistry often presents scenarios not explicitly encountered during training, demanding models to deduce properties and reactivity. Consequently, simply scaling up model size – increasing the number of parameters – proves insufficient; a model can memorize countless reactions without genuinely understanding why they occur, hindering its ability to predict novel outcomes or design new molecules. This limitation underscores the need for architectures that move beyond statistical correlation and embrace the principles of chemical intuition and mechanistic understanding.
Supervised fine-tuning, a common method for adapting large language models to specific tasks, frequently encounters the challenge of ‘catastrophic forgetting’. This phenomenon describes the tendency of a model, when trained on a new dataset, to abruptly and significantly lose previously acquired knowledge. Essentially, the model overwrites its existing understanding with the new information, diminishing its ability to generalize across a wider range of chemical problems. While effective at improving performance on the immediate task, this instability limits the model’s long-term adaptability and necessitates continuous retraining to maintain a broad competency, hindering its practical application in dynamic chemical research scenarios.
Simply increasing the size of large language models doesn’t guarantee improved performance in complex chemical reasoning; the field requires a fundamental shift in architectural design. Current models, despite their impressive ability to recognize patterns, often struggle with tasks demanding genuine understanding and problem-solving-a limitation stemming from their reliance on memorization rather than deductive logic. Recent work demonstrates that comparable tool-use capabilities can be achieved with significantly smaller models-ranging from 7 to 14 billion parameters-by prioritizing architectures that emulate the reasoning processes of chemists, suggesting that intelligent design, rather than sheer scale, holds the key to unlocking true progress in this domain.
![ChemCRAFT leverages a data-curation pipeline built on agentic trajectories within a chemical sandbox, followed by a two-stage training process-initial cold-start training with token-level loss for tool invocation, and subsequent refinement using Generalized Reinforcement Learning with Policy Optimization [latex]GRPO[/latex] to improve tool understanding.](https://arxiv.org/html/2601.17687v1/x1.png)
Orchestrating Intelligence: Offloading Complexity is Key
Cognitive decoupling addresses inherent limitations in large language models by enabling the offloading of computationally intensive or knowledge-dependent tasks to external, specialized tools. This approach bypasses the need for the model to internalize all required expertise, reducing parameter count and training data requirements. Rather than attempting to solve problems directly, the model learns to identify when a tool can assist and then formulates appropriate API calls, interprets the tool’s output, and integrates it into its reasoning process. This division of labor allows for increased efficiency and accuracy, particularly for tasks demanding specific domain knowledge or complex calculations, while simultaneously mitigating the challenges associated with scaling model size to encompass all possible expertise.
Multi-Agent Systems represent an architectural extension of cognitive decoupling, enabling language models to surpass inherent limitations by dynamically accessing and utilizing external Application Programming Interfaces (APIs). This approach moves beyond simple tool use to orchestrate interactions with a diverse suite of specialized tools, effectively creating a collaborative network for problem-solving. Specifically, Chemical Language Models benefit from this paradigm by gaining access to APIs for tasks such as molecular property calculation, reaction prediction, and database querying; this external access augments the model’s internal knowledge and allows it to perform complex chemical reasoning tasks that would otherwise be infeasible. The modular nature of these systems also facilitates easy integration of new tools and capabilities as they become available.
Tool-integrated reasoning centers on the construction of training datasets that explicitly incorporate interactions with external tools during the learning process, resulting in ‘Agentic Trajectories’ that detail a sequence of actions and observations. This approach enables the system to learn how to utilize tools to solve problems, rather than simply predicting outputs. Empirical results demonstrate that systems trained with this methodology achieve performance levels comparable to established commercial APIs. Furthermore, the inclusion of tool interactions significantly reduces the required input token length; specifically, a 65% reduction has been observed when contrasted with the SciToolAgent framework, indicating improved efficiency and reduced computational cost.
Refining the Narrative: Validation is Everything
Reflective Refinement improves reasoning performance by iteratively revising the model’s generated reasoning traces. This process leverages external tools to validate and, if necessary, correct intermediate steps in the reasoning process. Specifically, the model rewrites its initial trace with outputs verified by these tools, effectively incorporating external validation into its internal reasoning chain. This differs from simply using tools for a final answer check; instead, tool outputs become integral to the reasoning process itself, enabling correction and refinement of earlier steps and ultimately leading to more reliable conclusions.
Reinforcement Learning (RL) is employed to train the model in effective tool usage, mirroring the iterative problem-solving strategies of human chemists. Specifically, the Generative Reward-guided Policy Optimization (GRPO) algorithm facilitates the development of optimal policies for selecting and applying external tools. This approach allows the model to learn which tools are most appropriate for specific tasks and how to sequence their application to achieve desired outcomes. The RL framework provides a reward signal based on the verification of outputs generated by these tools, thereby guiding the model toward improved tool-use proficiency and enhanced reasoning capabilities.
The system’s performance in molecule understanding is significantly enhanced through the integration of cheminformatics libraries, notably RDKit, which facilitates molecular manipulation and calculation. Quantitative evaluation demonstrates a Molecule Understanding (MAE) score of 0.03 for Function-Group-Detection, exceeding the performance of Qwen2.5-32B, which achieved a MAE of 0.36. Furthermore, the system attained 100% accuracy in Ring-System-Detection, a substantial improvement over Gemini-2.5-Pro’s 87.5% accuracy on the same task. These results indicate a considerable advancement in the system’s ability to accurately interpret molecular structures.
ChemCRAFT: A Next-Generation Chemical Reasoning Framework
ChemCRAFT utilizes a principle termed ‘Cognitive Decoupling’ as a core architectural component. This approach separates the framework’s reasoning engine from the specific functionalities of external tools, enabling dynamic tool selection and integration. Rather than embedding tool usage directly into the model’s parameters, ChemCRAFT maintains a distinct planning and execution phase; the system first determines what needs to be done, then identifies and utilizes the appropriate external tool to accomplish that task. This decoupling allows for greater flexibility, scalability, and adaptability to new tools without requiring retraining of the core reasoning engine, facilitating a modular and extensible system for chemical reasoning.
ChemCRAFT’s training methodology utilizes ‘Agentic Trajectories,’ which involves defining a sequence of actions an agent should take to achieve a chemical reasoning task. This is coupled with an implementation of the ‘Hypothesis-Action-Observation Loop’ – the agent formulates a hypothesis, performs an action based on that hypothesis (e.g., applying a chemical transformation), observes the outcome, and then uses that observation to refine its subsequent hypotheses and actions. This iterative process facilitates robust self-correction, allowing the model to adapt and improve its performance over time without requiring explicit supervision for every scenario. The framework’s ability to learn from the consequences of its actions is central to its overall efficacy.
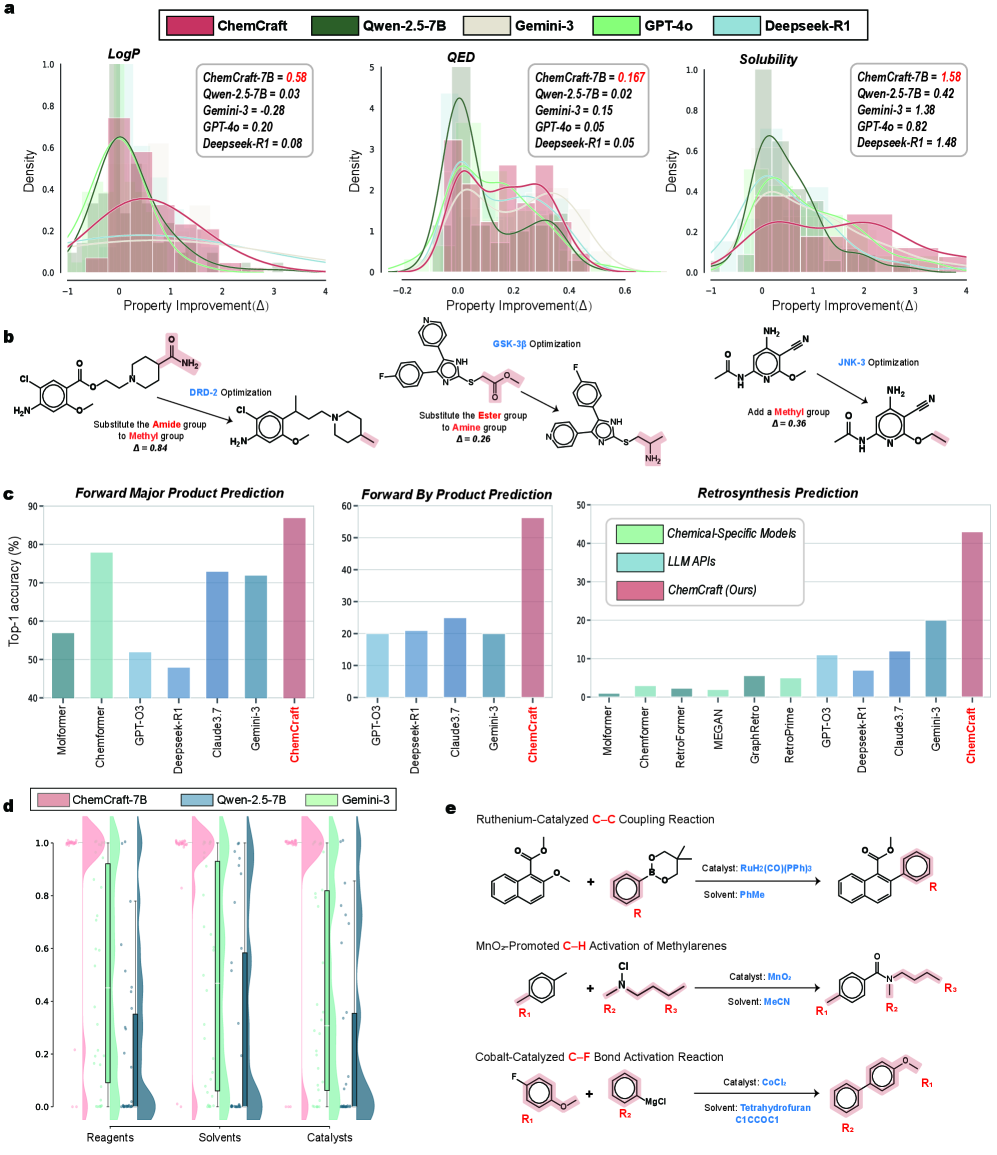
Performance of the ChemCRAFT framework was assessed using the ChemCoTBench benchmark suite, which evaluates capabilities in multi-step chemical reasoning and external tool utilization. Results indicate 97% SMILES Equivalence in molecule edit deletion tasks. Solubility optimization, measured as Δ, achieved a value of 1.58, representing a nearly four-fold improvement over Qwen (Δ=0.42) and exceeding the performance of Gemini-2.5-Pro (Δ=1.38). Furthermore, ChemCRAFT demonstrated retrosynthesis accuracy exceeding that of specialized models by a margin of 40% or greater.
The Future of Chemical AI: Beyond Current Limitations
The convergence of advanced language models with specialized external tools is revolutionizing chemical innovation. These models, adept at understanding and generating complex text, are now being paired with computational chemistry software, molecular databases, and robotic synthesis platforms. This synergy allows for the in silico prediction of molecular properties, the design of novel compounds with targeted characteristics, and the automated execution of experiments to validate these predictions. Crucially, robust refinement techniques – including feedback loops incorporating experimental data and iterative model retraining – are essential to overcome the inherent limitations of initial predictions and ensure the reliability of AI-driven discoveries. This combined approach is not merely accelerating research in areas like drug discovery and materials science, but is also opening doors to entirely new classes of compounds and materials previously inaccessible through traditional methods, promising solutions to global challenges in healthcare, energy, and sustainability.
The streamlined integration of artificial intelligence with established chemical practices hinges significantly on standardized molecular representations, and the Simplified Molecular Input Line Entry System – or SMILES – notation has emerged as a pivotal component. This text-based system allows complex molecular structures to be concisely encoded as strings, facilitating machine learning algorithms to ‘read’ and process chemical information effectively. Because SMILES is readily compatible with existing cheminformatics databases and software – tools already utilized for virtual screening, quantitative structure-activity relationship (QSAR) modeling, and other critical analyses – AI models can directly leverage decades of accumulated chemical knowledge. This avoids the need for laborious data reformatting and ensures that advancements in AI can be rapidly deployed within current workflows, accelerating research in areas like drug discovery and materials science by providing a common language for both chemists and computational systems.
Emerging frameworks like ChemCRAFT represent a significant leap forward in applying artificial intelligence to chemistry, poised to dramatically accelerate the pace of innovation across numerous fields. These systems aren’t simply predicting molecular properties; they are designed to actively design novel compounds with specific, desired characteristics – a capability with profound implications for drug discovery, materials science, and sustainable chemistry. By automating and optimizing the traditionally slow and iterative process of molecular design and synthesis, these AI-driven platforms hold the potential to address critical global challenges, from developing new pharmaceuticals to creating advanced materials for energy storage and environmental remediation. The core promise lies in a future where complex chemical problems are tackled not through exhaustive trial-and-error, but through intelligent, data-driven design, ushering in a new era of chemical AI that transcends the limitations of conventional methods.
The pursuit of elegant frameworks, as evidenced by ChemCRAFT’s decoupling of reasoning and computation, invariably invites eventual compromise. The system aims for expert-level problem solving, yet one anticipates the inevitable edge cases production will unearth – the unexpected reagent, the synthesis pathway that defies prediction. It’s a familiar pattern; complexity masked by clever design, only to be revealed by relentless real-world application. As Bertrand Russell observed, “The difficulty lies not so much in developing new ideas as in escaping from old ones.” ChemCRAFT, in its novelty, is simply building a new set of assumptions, destined to be tested – and likely broken – by the unforgiving logic of chemical reality. Tests are, after all, a form of faith, not certainty.
What’s Next?
The decoupling of reasoning and computation, as demonstrated by ChemCRAFT, feels less like an architectural triumph and more like a temporary reprieve. Every elegant abstraction eventually encounters the messy realities of production synthesis-failed reactions, unexpected byproducts, and the sheer cost of experimentation. The model’s current reliance on SMILES-GRPO, while effective, implicitly encodes limitations in representational power; the next iteration will invariably wrestle with the constraints of any chosen molecular encoding.
The reinforcement learning component, lauded for optimizing tool usage, will undoubtedly discover diminishing returns. Everything optimized will one day be optimized back, chasing a local maximum in a chemical space far more complex than any reward function can fully capture. The true test won’t be achieving expert-level performance on benchmark problems, but sustaining that performance in the face of novel challenges-the unforeseen edge cases that expose the brittleness of even the most sophisticated systems.
It’s tempting to envision increasingly autonomous chemical design, but the field should resist the allure of full automation. The value, in the long run, won’t be replacing chemists, but amplifying their intuition. This isn’t about building machines that think like chemists, but systems that efficiently surface the most promising hypotheses, allowing human expertise to focus on the genuinely creative leaps. We don’t refactor code-we resuscitate hope.
Original article: https://arxiv.org/pdf/2601.17687.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Invincible Season 4 Episode 4 Release Date, Time, Where to Watch
- How Martin Clunes has been supported by TV power player wife Philippa Braithwaite and their anti-nepo baby daughter after escaping a ‘rotten marriage’
- Beyond Accuracy: Gauging Trust in Human-AI Teams
- Gold Rate Forecast
- Clash Royale Balance Changes March 2026 — All Buffs, Nerfs & Reworks
- ‘Project Hail Mary’s Unexpected Post-Credits Scene Is Worth Sticking Around
- CookieRun: OvenSmash coupon codes and how to use them (March 2026)
- We talked to ‘Bachelorette’ Taylor Frankie Paul. Then reality hit pause on her TV career
- eFootball 2026 is bringing the v5.3.1 update: What to expect and what’s coming
- Only One Straw Hat Hasn’t Been Introduced In Netflix’s Live-Action One Piece
2026-01-27 22:42