Author: Denis Avetisyan
Researchers are leveraging the power of artificial intelligence to navigate complex chemical spaces and generate promising drug candidates.
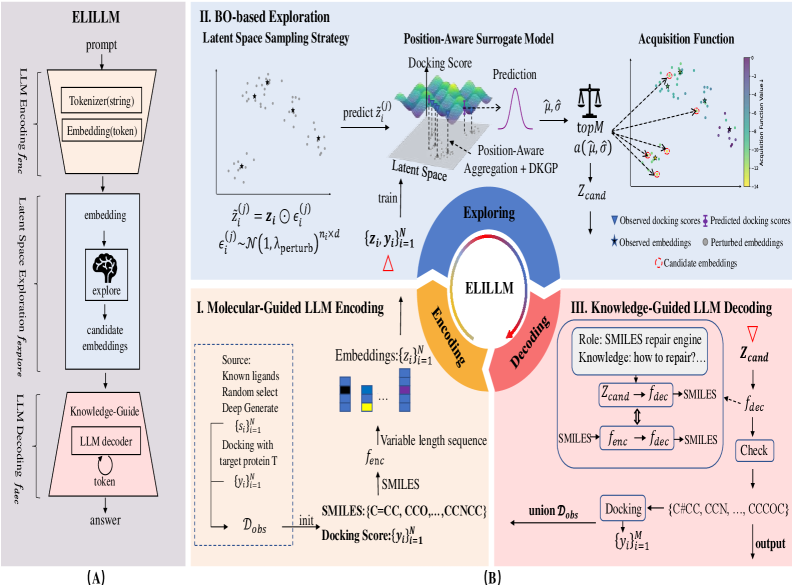
A novel framework, ELILLM, enhances large language models for structure-based drug design via Bayesian optimization of the latent space, improving the generation of high-affinity molecules.
While large language models exhibit strong representational and reasoning skills, their application to structure-based drug design is hampered by limited understanding of protein structures and unpredictable molecular generation. This work introduces a novel framework, ‘Empowering LLMs for Structure-Based Drug Design via Exploration-Augmented Latent Inference’ (ELILLM), which reimagines LLM generation as an encoding, latent space exploration, and decoding process. By systematically exploring the latent space with Bayesian optimization and employing knowledge-guided decoding, ELILLM enhances the generation of chemically valid and high-affinity molecules, as demonstrated on the CrossDocked2020 benchmark. Could this approach unlock a new era of AI-driven drug discovery, accelerating the identification of promising therapeutic candidates?
The Enduring Challenge of Molecular Representation
Structure-based drug design fundamentally hinges on the ability to forecast how strongly a molecule will bind to its target – a process often quantified by scoring functions like the Vina Docking Score. This score estimates the binding affinity, attempting to correlate a predicted energy state with actual biological activity. While computationally efficient, these scoring functions are approximations, and inaccuracies in predicting binding pose or accounting for solvation effects can lead to false positives or the overlooking of promising candidates. Consequently, a significant portion of drug discovery efforts are still dedicated to synthesizing and testing compounds predicted to have high scores, highlighting the persistent challenge of accurately translating computational predictions into real-world efficacy and the continuous need for improved scoring methodologies.
The effective representation of molecular structures is paramount in modern drug discovery, yet commonly employed methods face inherent limitations. While the Simplified Molecular Input Line Entry System (SMILES) string notation offers a concise and widely adopted approach to defining molecular structures, its sequential nature struggles to capture the full complexity of three-dimensional molecular geometry and intricate bonding patterns. This can be particularly problematic for advanced machine learning models – especially graph neural networks – which require a robust and nuanced understanding of molecular shape to accurately predict properties like binding affinity or solubility. Consequently, researchers are actively exploring alternative representations, including molecular graphs and more sophisticated string-based encodings, aiming to overcome the constraints of SMILES and unlock the potential of artificial intelligence in designing novel therapeutic compounds.
Initial forays into generative molecular design leveraged techniques like Variational Autoencoders and Autoregressive Models, offering a tantalizing glimpse of automated drug discovery. These early methods demonstrated the capacity to learn the underlying distributions of molecular properties and generate new structures. However, a significant hurdle quickly emerged: the generated compounds frequently lacked the desired high-affinity binding characteristics necessary for effective drug candidates. While capable of producing syntactically valid molecules, these models often struggled to navigate the complex chemical space and optimize for crucial pharmacological properties, resulting in compounds that were either unoriginal variations of known structures or entirely improbable and ineffective designs. This limitation spurred further research into more sophisticated generative architectures capable of balancing novelty with potency.
Harnessing Large Language Models for Rational Design
Large Language Models (LLMs), initially developed for natural language processing, exhibit substantial potential in molecular design due to their inherent capabilities in reasoning and generalization. These models are trained on extensive datasets, allowing them to identify patterns and relationships that can be extrapolated to novel scenarios – in this case, the relationships between molecular structure and properties. Unlike traditional computational methods which often rely on pre-defined rules or simulations, LLMs can learn directly from data, enabling the generation of molecules with desired characteristics without explicit programming of chemical rules. This capacity for pattern recognition and extrapolation allows LLMs to explore chemical space more efficiently and potentially discover compounds with optimized properties for specific applications, offering a complementary approach to conventional molecular design techniques.
Direct application of Large Language Models (LLMs) to molecular design is not possible with raw molecular data; therefore, Molecular Encoding is employed to transform complex molecular structures into a numerical representation suitable for LLM processing. This process creates a Latent Space, a multi-dimensional vector space where each point corresponds to a valid molecule. The dimensions of this space capture essential chemical features, allowing the LLM to navigate and explore the chemical space effectively. By representing molecules as vectors, LLMs can then perform operations like similarity searches, interpolation, and extrapolation to generate novel molecular structures based on learned patterns within the encoded data. The quality and dimensionality of the Latent Space directly impact the LLM’s ability to generate valid and desirable molecules.
Knowledge-Guided Decoding enhances molecular generation by Large Language Models through the integration of established chemical principles. This is achieved by conditioning the LLM’s decoding process – the step where it predicts subsequent molecular elements – on pre-existing chemical knowledge, often represented as rules or constraints. Specifically, this involves biasing the probability distribution of potential molecular fragments towards those that adhere to validated chemical properties, such as valency rules or desirable functional group arrangements. The result is a significant increase in the proportion of generated molecules that are chemically valid and possess characteristics aligned with specific design objectives, thereby reducing the need for extensive post-generation filtering and improving the efficiency of the molecular discovery process.
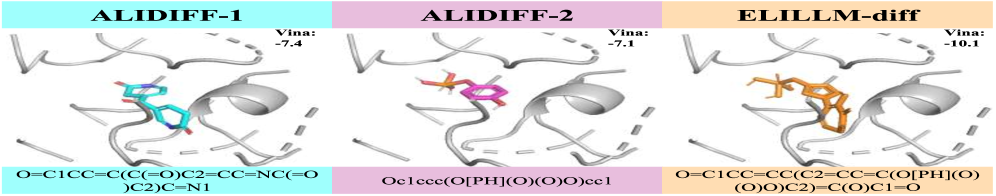
![ALIDIFF and ELILLM-diff successfully generated ligands tailored to the [latex]3nfb[/latex] protein pocket.](https://arxiv.org/html/2601.15333v1/x5.png)
ELILLM: An Exploration-Augmented Framework for Discovery
The Exploration-Augmented Latent Inference (ELILLM) framework addresses molecule generation by integrating Large Language Models (LLMs) with Bayesian Optimization. This synergistic approach leverages the generative capabilities of LLMs within a latent space, while Bayesian Optimization provides a statistically principled method for efficiently exploring this space. Unlike traditional generative models, ELILLM doesn’t rely on random sampling; instead, Bayesian Optimization intelligently proposes molecule candidates based on predicted properties, maximizing the probability of discovering high-performing compounds with fewer iterations. This targeted exploration enhances the efficiency of the molecule generation process and reduces the computational cost associated with screening vast chemical spaces.
The ELILLM framework utilizes a Position-Aware Surrogate Model to enhance molecule generation efficiency. This model operates within the latent space, predicting binding affinity for generated compounds without requiring computationally expensive 3D structural evaluations. By learning the relationship between latent vector positions and predicted binding affinities, the surrogate model functions as a guide during the Bayesian Optimization process. This allows ELILLM to prioritize exploration of latent regions likely to yield molecules with improved binding characteristics, effectively reducing the search space and accelerating the identification of promising candidates.
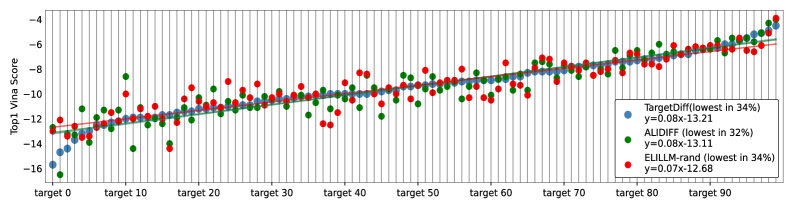
Quantitative analysis demonstrates that the ELILLM framework achieves statistically significant improvements in predicted binding affinity compared to the ALIDiff baseline. Specifically, ELILLM exhibits a -4.59% improvement when evaluating the top candidate molecule, and progressively larger improvements when considering the top 5 (-5.88%), top 10 (-6.69%), and top 20 (-7.77%) candidates. These improvements were determined to be statistically significant based on a Wilcoxon signed-rank test, yielding a p-value of less than 0.05.
ELILLM builds upon the principles of Structure-Based Drug Design by leveraging computational methods to predict the interaction between potential drug candidates and target proteins. This framework moves beyond traditional methods through the integration of Bayesian Optimization and a Position-Aware Surrogate Model, enabling efficient exploration of the chemical space and prediction of binding affinity within a latent representation. The resulting system facilitates the de novo generation of molecules, offering a scalable approach to identifying novel compounds with demonstrably improved properties, as evidenced by statistically significant improvements in binding affinity compared to existing methods like ALIDiff. This computational efficiency and performance improvement positions ELILLM as a robust solution for accelerating drug discovery pipelines.
Expanding the Horizon: Impact and Future Directions
The efficiency with which ELILLM navigates chemical space represents a significant leap toward accelerated materials discovery and therapeutic development. By intelligently sampling molecular configurations, the framework bypasses the limitations of traditional methods that often require exhaustive and computationally expensive searches. This capability allows researchers to pinpoint promising candidates – novel molecules with desired properties – far more rapidly than previously possible. The potential impact extends to diverse fields, from designing new catalysts and polymers with enhanced performance to identifying drug leads for currently untreatable diseases. Ultimately, ELILLM doesn’t just generate molecules; it unlocks access to a vast, previously inaccessible realm of chemical innovation, promising a future where materials and medicines are discovered with unprecedented speed and precision.
The efficiency of ELILLM extends beyond individual molecule design, offering a pathway to dramatically accelerate drug discovery through high-throughput screening. By rapidly generating and evaluating a vast chemical space, the framework enables researchers to identify promising candidate molecules with desired properties at an unprecedented rate. This scalability isn’t simply about processing more data; it’s about intelligently navigating the complex landscape of molecular possibilities, prioritizing compounds likely to succeed in later stages of development. Consequently, the time and resources traditionally required for iterative synthesis and testing can be significantly reduced, potentially bringing novel therapeutics to market faster and more cost-effectively. The ability to optimize molecular properties in silico, before physical synthesis, represents a paradigm shift in how researchers approach the challenges of modern drug design.
Ongoing development prioritizes enhancing the precision of the Position-Aware Surrogate Model, a crucial component for efficiently navigating the vast landscape of potential molecular structures. Researchers aim to move beyond simple positional encoding, integrating more nuanced understandings of how molecular context influences properties and reactivity. Simultaneously, the Knowledge-Guided Decoding process will be expanded to incorporate increasingly complex chemical rules and constraints, moving from basic validity checks to predictive assessments of synthetic accessibility and pharmacological behavior. These combined improvements promise to not only accelerate the discovery of promising candidate molecules but also to increase the likelihood of successfully translating in silico predictions into tangible, real-world applications, ultimately streamlining the development of novel therapeutics and advanced materials.
The pursuit of novel molecular structures, as detailed in the exploration of ELILLM, benefits from a focused reduction of complexity. The framework elegantly navigates the latent space of Large Language Models, optimizing for high-affinity molecules through Bayesian optimization-a process akin to distilling information to its essential form. As Bertrand Russell observed, “The point of education is not to increase the amount of information, but to create the capacity for critical thinking.” Similarly, ELILLM doesn’t simply generate possibilities; it intelligently searches and refines within the model’s knowledge, embodying the principle that true advancement lies not in accumulation, but in skillful subtraction and targeted exploration of the relevant solution space.
The Simplest Path Forward
The presented framework, while demonstrating a measurable improvement in molecular generation, merely addresses a symptom. The underlying complexity of structure-based drug design-a vast chemical space coupled with imperfect scoring functions-remains stubbornly intact. The true measure of progress will not be in increasingly intricate algorithms, but in methods that reduce the need for exhaustive search. A system that requires optimization, however clever, has already conceded a fundamental failure of predictive power.
Future efforts should prioritize the distillation of biological truth into simpler, more readily exploitable models. The latent space exploration, while valuable, functions as a sophisticated form of trial and error. The goal is not to refine the error, but to eliminate its necessity. One anticipates, perhaps optimistically, a shift towards principles-based design, where molecular properties emerge from fundamental physical constraints rather than being statistically inferred.
Clarity is, after all, a courtesy. The proliferation of parameters and layers, while impressive from an engineering standpoint, obscures the core challenge: understanding-and ultimately, predicting-molecular behavior. A successful theory will not generate more data; it will demand less.
Original article: https://arxiv.org/pdf/2601.15333.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-01-24 02:08