Author: Denis Avetisyan
Researchers have developed a novel deep learning framework to more accurately predict how small molecules interact with proteins, a crucial step in drug discovery and understanding biological processes.
![The study demonstrates that accurate prediction of protein-ligand binding affinity relies on precise consideration of functional group interactions-specifically, the affinity between carbonyl and pyridine groups-as neglecting these constraints leads to erroneous predictions of weak interactions, such as those incorrectly posited between carbon and oxygen atoms [latex] (C \leftrightarrow O) [/latex] instead of the correct interaction between carbon and nitrogen [latex] (C \leftrightarrow N) [/latex].](https://arxiv.org/html/2602.05479v1/x1.png)
Phi-Former leverages hierarchical modeling and pre-training to improve prediction of compound-protein binding affinity by representing interactions at both atomic and motif levels.
Predicting how compounds interact with proteins remains a significant bottleneck in drug discovery, despite recent advances in deep learning. This work introduces ‘Phi-Former: A Pairwise Hierarchical Approach for Compound-Protein Interactions Prediction’, a novel framework that addresses limitations of current models by explicitly incorporating the biological relevance of molecular motifs. Phi-Former learns hierarchical representations and utilizes a pairwise pre-training strategy to model interactions across atom, motif, and atom-motif levels, improving prediction accuracy and interpretability. Could this approach unlock more rational drug design strategies and accelerate the development of precision medicines?
The Intractable Challenge of Molecular Binding Prediction
The pursuit of novel therapeutics hinges on the ability to accurately forecast how strongly a potential drug candidate will bind to its target protein; this binding affinity is a primary determinant of a drug’s efficacy. However, predicting this interaction strength remains a formidable obstacle in drug discovery. Initial screening often identifies compounds that appear promising, but a substantial proportion fail in later stages due to unexpectedly weak or nonexistent binding in a biological environment. This disconnect stems from the inherent complexity of molecular interactions – factors such as protein flexibility, solvent effects, and entropic contributions are difficult to model with precision. Consequently, researchers expend considerable resources on compounds with low probability of success, driving up the cost and lengthening the timeline for bringing new medicines to patients. Improving the reliability of binding affinity prediction is therefore paramount to streamlining the drug development process and increasing the likelihood of clinical success.
The pharmaceutical industry faces a substantial hurdle in bringing novel therapeutics to market, largely due to the inherent difficulty in predicting how strongly a potential drug will bind to its target protein – a measure known as binding affinity. Traditional drug discovery methods, often relying on high-throughput screening and simplified models of molecular interaction, frequently misjudge this critical factor. This leads to a cascade of problems, as compounds exhibiting promising in vitro activity fail to translate into effective treatments during clinical trials. The complexity arises from the subtle interplay of forces – electrostatic, hydrophobic, van der Waals – governing these interactions, and the difficulty in accurately modeling the protein’s flexibility and the solvent environment. Consequently, a significant percentage of drug candidates are rejected at late stages of development, representing a substantial financial loss and delaying the availability of needed medications.
Despite advancements in computational power and algorithm design, current models attempting to predict the strength of compound-protein interactions – known as binding affinity – still face limitations. These models, which rely on analyzing protein and compound sequences or three-dimensional structures, often struggle with the inherent complexity of biomolecular recognition. Subtle changes in protein conformation, the flexibility of the binding pocket, and the influence of solvent effects are frequently underestimated or simplified. Consequently, predictions can deviate significantly from experimental values, hindering the efficient identification of promising drug candidates. Ongoing research focuses on incorporating more sophisticated physics-based simulations, machine learning techniques trained on extensive datasets, and improved representations of protein dynamics to achieve greater predictive accuracy and ultimately accelerate the drug discovery process.
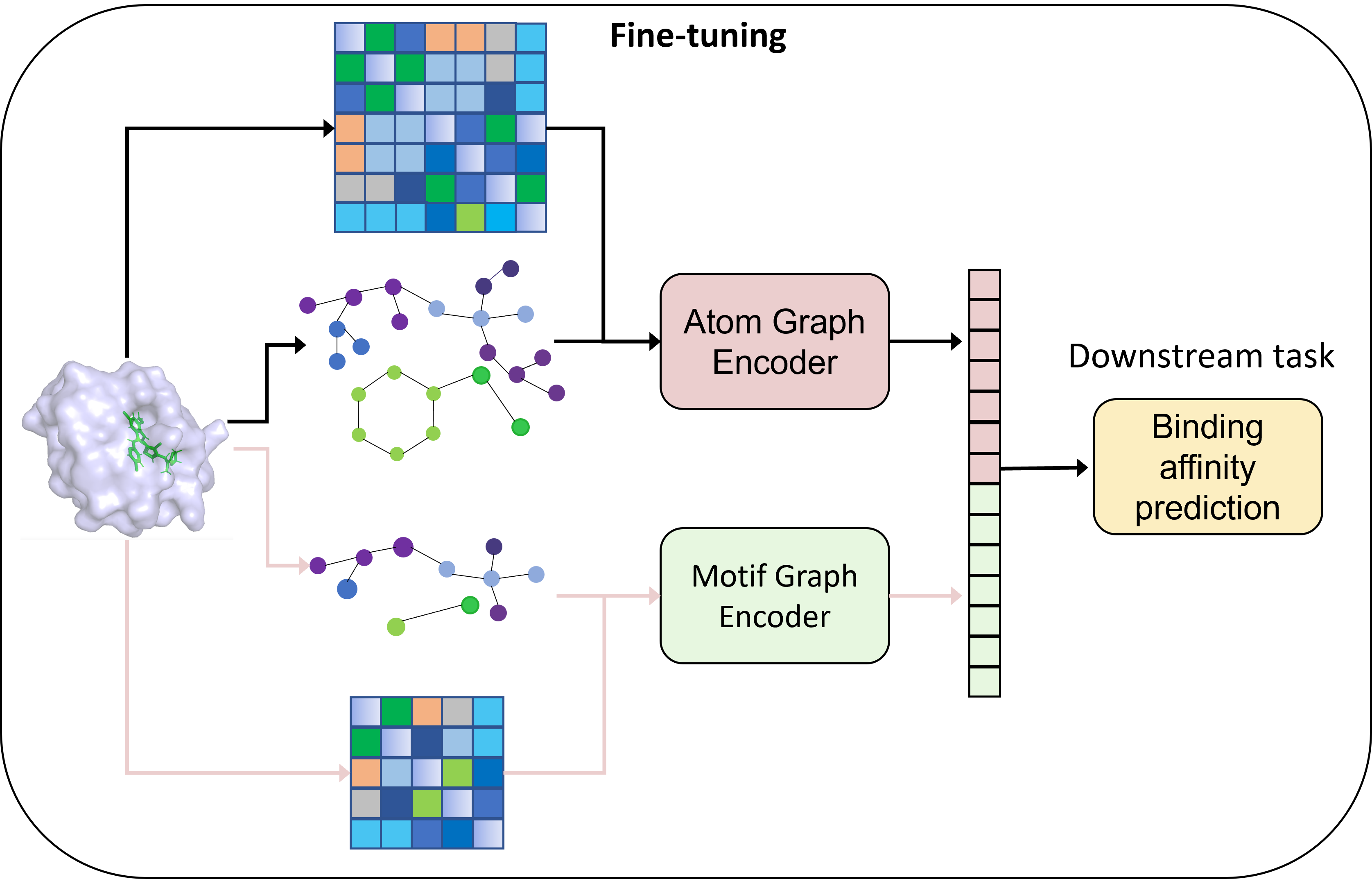
Phi-former: A Hierarchical Representation of Molecular Interactions
Phi-former employs a pairwise hierarchical interaction representation learning framework built upon Graph Transformers to model chemical properties. This approach represents molecules as graphs where nodes are atoms and edges represent interactions. The Graph Transformer architecture allows the model to learn relationships between these atoms by iteratively updating node representations based on their connections. Pairwise interactions are explicitly modeled to capture the influence of each atom on its neighbors, and the hierarchical structure enables representation learning at multiple levels of abstraction, allowing the model to capture both local and global molecular features. This differs from traditional graph neural networks by explicitly focusing on pairwise interactions and leveraging the attention mechanism within the Transformer to weigh the importance of each interaction during representation learning.
Phi-former employs a hierarchical modeling strategy to represent molecular structures by progressively abstracting information. The model initiates representation at the atomic level, constructing a graph where nodes are individual atoms and edges denote bonding or spatial relationships. This atom-level graph is then used to identify and represent recurring substructures, termed motifs. These motifs, representing higher-level building blocks within the molecule, are themselves organized into a graph, forming the subsequent layer of abstraction. This iterative process of graph construction at increasing levels of abstraction – from atoms to motifs and potentially higher-order assemblies – allows the model to capture complex molecular features and relationships in a computationally efficient manner, reducing the complexity of direct atom-to-atom interaction calculations.
Spatial Positional Encoding (SPE) is a key component of the Phi-former model, addressing the limitations of standard Graph Transformers in representing 3D molecular geometry. Unlike approaches that treat nodes as unordered sets, SPE incorporates the [latex]x, y, z[/latex] coordinates of each atom directly into the positional encoding vector. This is achieved by calculating distances between atoms and embedding these distances as features within the encoding. By explicitly providing the model with spatial information, SPE enables it to differentiate between molecules with the same connectivity but different 3D conformations, which is critical for accurately predicting molecular properties and interactions dependent on spatial arrangement, such as binding affinity and reaction rates. The encoding is added to the initial node embeddings, allowing the Transformer layers to effectively utilize 3D coordinate data during message passing and attention mechanisms.
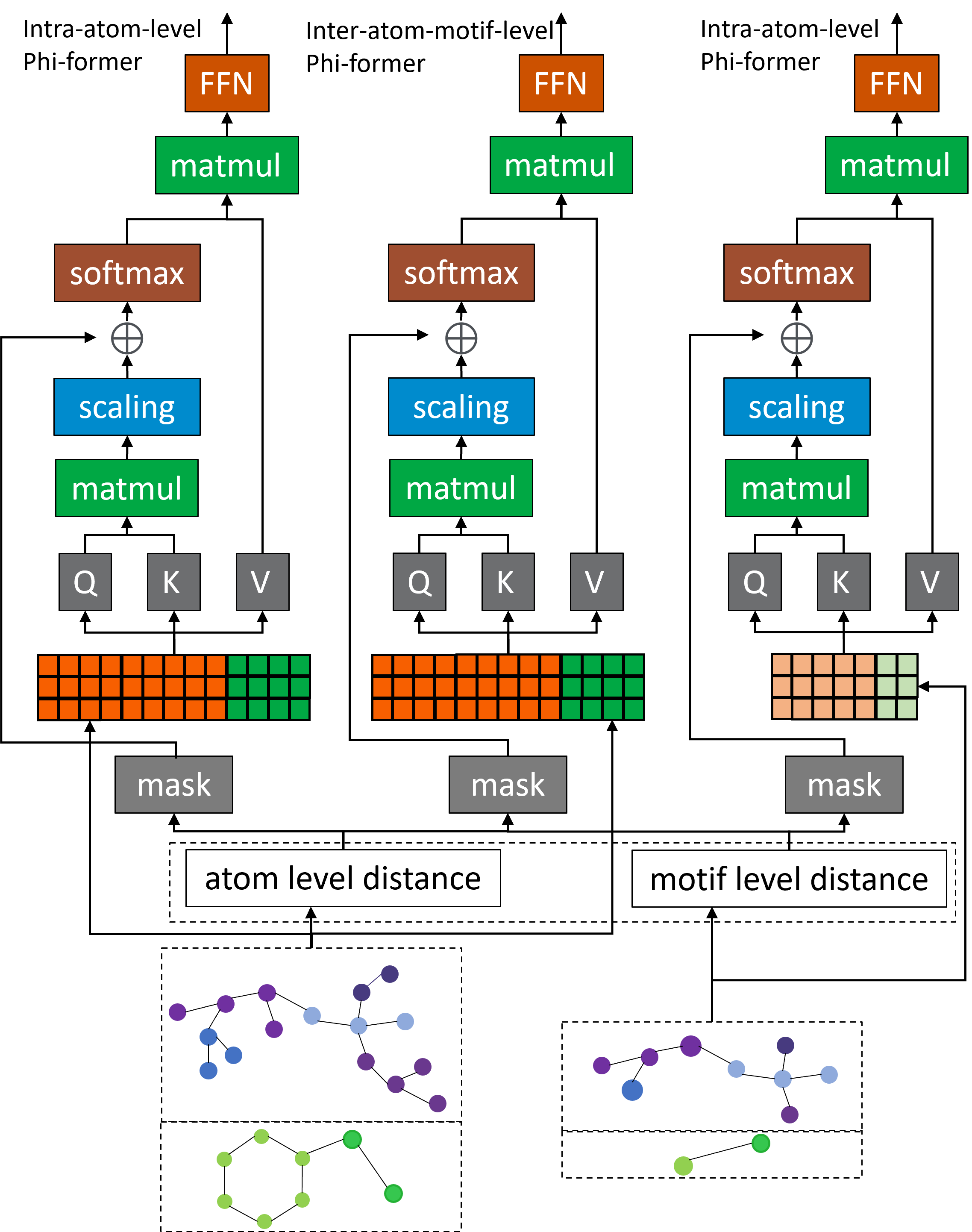
Multi-Level Optimization Through Hierarchical Loss Functions
Phi-former utilizes a dual-loss optimization strategy comprised of Intra-Loss and Inter-Loss functions to facilitate learning at multiple hierarchical levels within the compound-protein complex. The Intra-Loss function focuses on optimizing representations within individual levels of the hierarchy, ensuring accurate modeling of local interactions. Simultaneously, the Inter-Loss function optimizes interactions between these hierarchical levels, enabling the model to capture global dependencies and relationships. This combined approach allows for a more comprehensive understanding of the complex, leading to improved predictive performance compared to methods that address only local or global features in isolation.
The implementation of both intra-loss and inter-loss functions within the Phi-former model facilitates the capture of both localized and broad-scale relationships within the compound-protein complex. Intra-loss focuses on optimizing interactions at the individual residue or atom level, ensuring accurate representation of direct binding contacts. Conversely, the inter-loss component optimizes interactions between spatially distant residues or atoms, thereby modeling the allosteric effects and long-range dependencies critical for accurate binding affinity prediction. This combined approach allows the model to simultaneously refine local precision and global context, improving the overall understanding of the complex’s structural and energetic properties.
Evaluation on the PDBBind 2019 dataset demonstrates that the model outperforms existing compound-protein interaction prediction methods. Specifically, the model achieved a Root Mean Squared Error (RMSE) of 1.159 and a Pearson correlation coefficient of 0.846. These results represent an improvement over the performance metrics reported for MONN, TankBind, OnionNet, IGN, and SS-GNN when evaluated on the same dataset, indicating a higher degree of accuracy in predicting binding affinities.
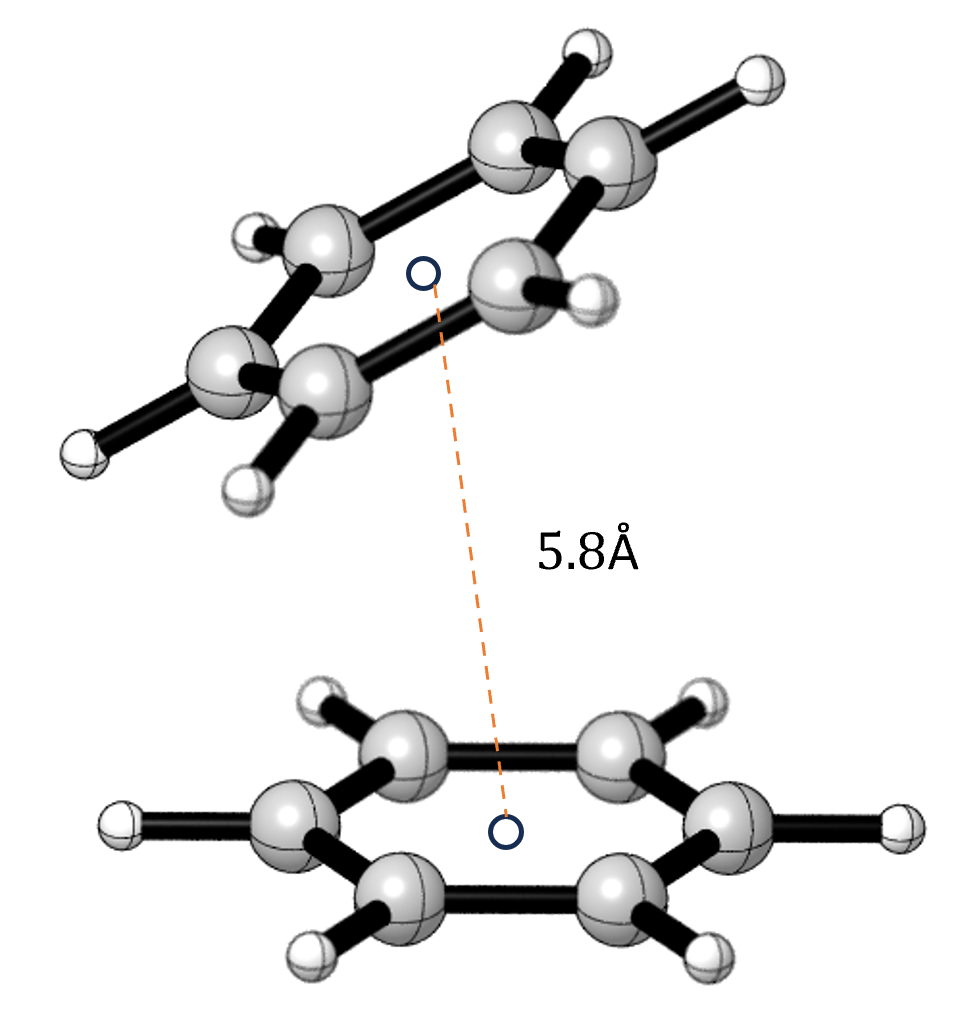
Dissecting the Role of Non-Covalent Interactions in Binding
Phi-former distinguishes itself by meticulously modeling interactions at various levels of detail, moving beyond simplified representations of molecular forces. This approach allows for a nuanced understanding of how subtle, non-covalent interactions-particularly aromatic π-π stacking-contribute to the stability and function of biomolecules. By explicitly accounting for these forces, which arise from the overlap of electron clouds in aromatic rings, Phi-former can accurately predict binding affinities and structural properties. The model doesn’t merely register the presence of a π-π interaction, but quantifies its strength and orientation within the protein-ligand complex, offering researchers a powerful tool to dissect the molecular basis of binding and design more effective therapeutic compounds.
The efficacy of a drug hinges not only on its primary chemical structure but also on the subtle, yet critical, non-covalent interactions it forms within the body. These interactions – encompassing hydrogen bonds, van der Waals forces, and hydrophobic effects – dictate how a drug molecule binds to its target protein, influencing both affinity and selectivity. A thorough understanding of these forces allows researchers to move beyond trial-and-error approaches to drug development, enabling rational drug design where molecules are specifically engineered to maximize favorable interactions with the target while minimizing off-target effects. Optimization of drug candidates then focuses on fine-tuning these interactions to enhance potency, improve bioavailability, and ultimately, increase the probability of clinical success, significantly reducing the time and resources traditionally associated with bringing new therapies to patients.
The predictive capabilities of Phi-former represent a substantial advancement in computational drug discovery, as demonstrated by its performance on the PDBBind 2019 dataset. Achieving a root mean squared error (RMSE) of 1.159 and a Pearson correlation coefficient of 0.846 signifies a high degree of accuracy in predicting protein-ligand binding affinities. This level of precision allows researchers to virtually screen vast libraries of compounds, identifying promising drug candidates with greater confidence and efficiency. Consequently, the reliance on costly and time-consuming physical experiments is diminished, potentially accelerating the drug development timeline and significantly reducing overall research and development expenditures. The model’s ability to accurately forecast binding strength offers a powerful tool for rational drug design, enabling the optimization of drug candidates before synthesis and testing, and ultimately increasing the probability of successful therapeutic outcomes.
The Phi-Former framework, with its emphasis on hierarchical modeling of compound-protein interactions, resonates with a pursuit of fundamental correctness. The authors’ focus on representing interactions at both atomic and motif levels exemplifies a desire to move beyond mere empirical observation toward a more mathematically grounded understanding of binding affinity. This aligns with Kernighan’s observation that “Debugging is twice as hard as writing the code in the first place. Therefore, if you write the code as cleverly as possible, you are, by definition, not smart enough to debug it.” While not directly about debugging, the Phi-Former’s rigorous approach to representation learning mirrors a desire for provable, rather than simply functional, models, reducing the need for post-hoc adjustments and increasing confidence in predictions. The method’s pre-training strategy seeks to establish a robust foundation, a sort of ‘correctness by construction’, before tackling the complexities of specific interactions.
What Remains to be Proven?
The Phi-Former framework, while demonstrating improved predictive capability, merely shifts the locus of uncertainty. The hierarchical modeling, predicated on atomic and motif levels, implicitly assumes these representations are sufficient. A formal demonstration-a provable link between structural features and interaction probability-remains elusive. Current metrics, even with pre-training strategies, are still approximations of binding affinity, not rigorous derivations.
Future work must move beyond empirical observation. The reliance on graph neural networks, powerful as they are, lacks inherent mathematical guarantees. Defining a complete and consistent set of axioms for compound-protein interactions-a formal language describing binding events-is paramount. Only then can one move from ‘works well’ to ‘is correct’.
The ultimate challenge isn’t simply predicting whether a compound interacts, but why. A reductionist approach-deconstructing interaction forces into fundamental, quantifiable components-offers a pathway. Until such a framework exists, the prediction of compound-protein interactions will remain a sophisticated form of pattern recognition, not a true scientific explanation.
Original article: https://arxiv.org/pdf/2602.05479.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-07 09:37