Author: Denis Avetisyan
Generative AI is rapidly transforming enzyme engineering, offering new routes to create optimized biocatalysts for industrial applications.
This review examines the latest advances in using generative models – including diffusion and protein language models – for de novo enzyme design and performance enhancement.
Despite decades of limited success in de novo enzyme design, recent advances in artificial intelligence are rapidly transforming the field of biocatalysis. This review, ‘Generative AI for Enzyme Design and Biocatalysis’, provides a comprehensive overview of emerging generative models – encompassing both sequence and structural generators – now being employed to create and optimize enzymes for industrial applications. We highlight models with experimental validation, assess current limitations, and argue that generative AI has reached a maturity enabling significant progress in enzyme engineering. Can wider adoption of these models, coupled with experimental feedback, accelerate the development of next-generation biocatalysts and guide the evolution of even more powerful design tools?
Beyond Evolutionary Constraints: Designing Enzymes Anew
Enzyme engineering has historically centered on the iterative improvement of naturally occurring proteins, a strategy that, while successful in many instances, is fundamentally limited by the constraints of evolution. Existing enzymes are the product of billions of years of adaptation to biological roles, not necessarily optimal biocatalysts for industrial applications. This evolutionary baggage often manifests as performance plateaus, where further modification yields diminishing returns. The inherent structure and amino acid composition, shaped by natural selection for stability and regulation within a living organism, can hinder the achievement of desired traits like increased activity, altered substrate specificity, or enhanced stability under harsh conditions. Consequently, researchers increasingly recognize that surpassing these limitations requires venturing beyond the confines of existing enzymes and embracing entirely novel protein designs.
The creation of entirely new enzymes, designed de novo, represents a significant departure from traditional protein engineering and unlocks the potential for biocatalysis beyond the constraints of naturally evolved systems. Unlike modifying existing enzymes, which often encounters performance limits due to inherent evolutionary history, de novo design allows researchers to build catalysts with precisely tailored properties – optimizing for activity, stability, and selectivity towards specific reactions. This approach facilitates the development of enzymes suited to industrial applications where existing biocatalysts fall short, offering solutions for sustainable chemical synthesis, bioremediation, and the production of novel materials. By circumventing the limitations of natural evolution, scientists can engineer enzymes that operate under extreme conditions, utilize non-natural substrates, or catalyze reactions not found in nature, pushing the boundaries of what is possible with biological catalysts.
The challenge of crafting enzymes from scratch, or de novo design, resides in the sheer immensity of possibilities within protein structure. Each protein is defined by its amino acid sequence, and the number of potential sequences is effectively infinite. Researchers must navigate this vast ‘sequence space’ – a combinatorial explosion of possibilities – to identify those that will fold into a stable, functional three-dimensional structure. Accurate prediction of this structure from the sequence remains a significant hurdle, as even minor changes can dramatically alter a protein’s catalytic activity. Computational methods, coupled with advances in structural biology, are increasingly employed to model protein folding and assess the likelihood of a designed sequence yielding the desired enzymatic function, but validating these predictions through laboratory experiments remains crucial to realizing the full potential of de novo enzyme creation.

Generative AI: A Paradigm Shift in Protein Creation
Generative artificial intelligence (AI) is significantly impacting protein design through the analysis of extensive biological datasets. These models learn the complex relationships between amino acid sequences, three-dimensional structures, and functional properties by identifying patterns within these datasets. Recent data indicates a substantial increase in the success rate of protein design campaigns utilizing generative AI; the number of successful designs has effectively doubled over the past two years. This improvement is attributed to the models’ increasing ability to predict stable and functional protein structures, accelerating the process of de novo protein creation and optimization.
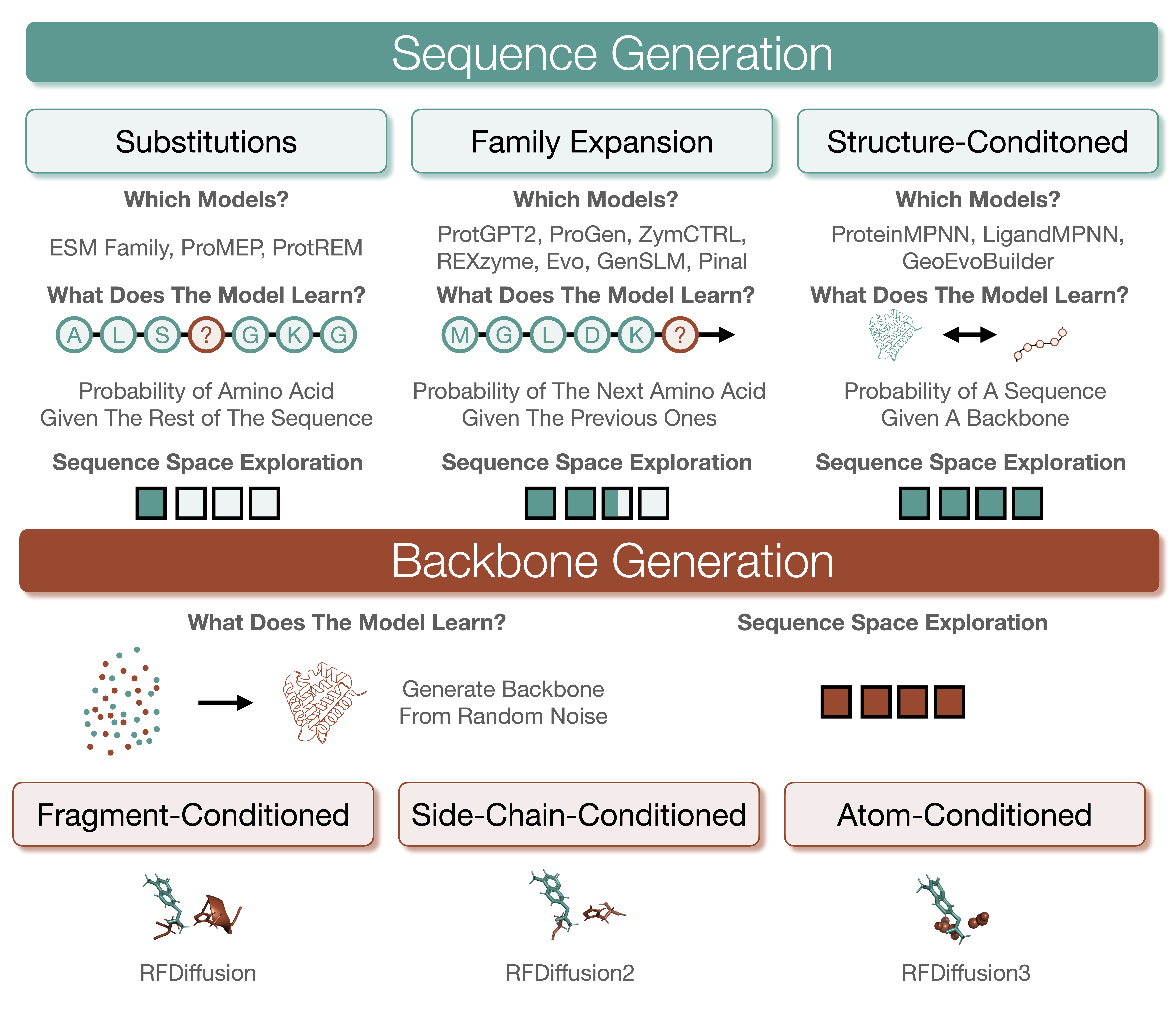
Sequence-generating models utilize deep learning architectures, most notably the Transformer, to design de novo protein sequences. These models are trained on extensive datasets of known protein sequences and structures, enabling them to learn the statistical relationships between amino acids and predict probable sequences. By defining specific characteristics – such as binding affinity, stability, or enzymatic activity – as target parameters, the models can generate novel amino acid sequences optimized to fulfill those criteria. The Transformer architecture’s attention mechanism allows the model to consider long-range dependencies within the sequence, improving the accuracy and functionality of the generated proteins. These generated sequences are not simply rearrangements of existing proteins but represent genuinely new protein designs.
The integration of physicochemical priors into generative AI protein design models enhances their ability to create viable protein structures. These priors, representing established principles of physics and chemistry – including electrostatic interactions, hydrophobic effects, and steric constraints – function as guiding parameters during sequence generation. By incorporating these constraints, the models are less likely to propose sequences that would result in physically implausible or unstable protein folds. Specifically, priors can be implemented as penalty terms within the model’s loss function, discouraging sequences that violate fundamental biophysical rules, and thereby increasing the probability of generating functional proteins with desired properties.
Autoregressive sequence models function by iteratively predicting the probability distribution of the next amino acid in a protein sequence, conditional on the preceding amino acids. This process begins with an initial sequence, which can be a short seed or a randomly generated string, and the model then predicts the most likely subsequent amino acid based on its training data. The predicted amino acid is appended to the sequence, and this extended sequence serves as the input for predicting the next amino acid, continuing until a complete protein sequence of a specified length is generated. This iterative, sequential prediction allows for the de novo creation of entirely new protein sequences, differing from those observed in naturally occurring proteins, by sampling from the predicted probability distributions.
Structure-Conditioned Design: Bridging Sequence and Geometry
Structure-conditioned protein design models, including ProteinMPNN and LigandMPNN, move beyond traditional sequence generation by directly integrating three-dimensional structural information into the design process. These models operate by conditioning the sequence generation on a target structure, or a template structure with a defined active site. This allows for the prediction of amino acid sequences compatible with a specific conformation, effectively focusing design efforts on optimizing residues within and surrounding the active site for enhanced binding affinity or catalytic activity. The incorporation of structural restraints significantly reduces the search space for viable sequences, enabling the design of proteins with pre-defined structural features and improved functional properties compared to purely sequence-based methods.
Backbone-generating models, prominently including RFDiffusion, initiate de novo protein design by directly predicting three-dimensional protein structures. These models utilize diffusion processes to iteratively refine structural proposals, ultimately producing a complete protein scaffold without relying on existing sequence or structural templates. The resulting structures then serve as a fixed framework onto which amino acid sequences are computationally optimized, effectively decoupling structure prediction from sequence generation and allowing for targeted sequence design to fit a predetermined conformation. This approach contrasts with sequence-based methods and offers a pathway to explore novel protein folds and architectures.
Linking protein sequence and structure during the design process enables the creation of proteins with predictably enhanced stability and function. Traditionally, sequence design focused primarily on amino acid composition for desired activity, often resulting in unstable proteins. Structure-conditioned models address this by incorporating spatial constraints and energetic considerations during sequence generation. This co-design approach allows for the optimization of residue placement to maximize favorable interactions within the protein fold, leading to increased thermodynamic stability. Consequently, these designed proteins demonstrate improved resistance to denaturation and aggregation, crucial for applications in targeted biocatalysis where sustained enzymatic activity under varying conditions is required. The ability to rationally engineer both sequence and structure expands the range of achievable protein functions and improves the efficiency of enzymatic processes.
Multiple sequence alignments (MSAs) are employed to refine computationally generated protein sequences by capitalizing on evolutionary data. MSAs identify conserved residues and patterns across homologous protein families, providing statistical measures of residue importance and permissible variations. These alignments are used to score and rank generated sequences, favoring those that align with known functional sequences and exhibit high conservation scores. Furthermore, MSAs can inform probability distributions used during sequence sampling, biasing the generation process towards sequences predicted to be stable and functional based on evolutionary precedent, thereby improving the likelihood of successful protein folding and activity.
Validating AI-Generated Enzymes: From Prediction to Performance
The ESM (Evolutionary Scale Modeling) family of models, developed by Meta AI, currently represents the state-of-the-art in protein sequence modeling and are increasingly utilized within the field of enzyme engineering. These models leverage large datasets of naturally occurring protein sequences to learn the relationships between amino acid sequences and protein structure and function. Specifically, ESM models are based on the Transformer architecture and trained using a masked language modeling objective, allowing them to predict missing amino acids within a sequence. This capability is crucial for de novo enzyme design, as it enables the prediction of stable and functional protein sequences without relying on known structural templates. Variations within the ESM family, such as ESM-1b and ESMFold, offer differing trade-offs between computational cost and predictive accuracy, catering to a range of enzyme engineering applications, from initial sequence generation to detailed structural refinement.
ProteinGym is a standardized benchmark suite used to quantitatively assess the accuracy of protein sequence models, specifically evaluating the correlation between amino acid probabilities predicted by the model and experimentally determined mutational effects. The platform utilizes a large, diverse set of protein backbones and measures the Pearson correlation coefficient between predicted and observed changes in protein stability upon mutation. High correlation scores indicate the model accurately captures the biophysical principles governing protein folding and stability, providing confidence in its predictions for enzyme design. This validation is crucial as it links in silico predictions to experimental outcomes, allowing researchers to refine and improve the predictive power of AI-driven enzyme engineering pipelines.
Validation of AI-generated enzymes is critical to confirm predicted functionality prior to application. Recent studies demonstrate that designs generated using sequence models can achieve catalytic efficiencies of [latex]2.2 \times 10^5[/latex] M-1 s-1, which is comparable to the median efficiencies observed in naturally occurring serine hydrolases. This level of performance indicates a strong correlation between in silico predictions and experimental outcomes, supporting the viability of AI-driven enzyme engineering for applications requiring specific catalytic activity.
Semi-Rational Design leverages the predictive capabilities of artificial intelligence in conjunction with directed evolution techniques to optimize enzyme properties beyond what either method could achieve independently. In one documented case, applying this approach to the Tobacco Etch Virus (TEV) protease resulted in a 26-fold increase in catalytic activity compared to the wild-type enzyme. Furthermore, the engineered TEV protease exhibited a 40°C increase in melting temperature, indicating improved thermostability. This combined methodology not only enhances existing enzyme function but also expands the accessible design space for novel protein engineering applications by iteratively refining AI-generated designs through experimental validation and subsequent model updates.
The Future of Enzyme Engineering: Tailoring Biocatalysts for a Sustainable Future
The field of biocatalysis stands on the cusp of a significant transformation, driven by the synergistic pairing of generative artificial intelligence and protein engineering. This innovative approach moves beyond traditional enzyme modification, allowing for the de novo design of enzymes precisely tailored to specific industrial needs. By leveraging AI algorithms to predict protein structures and functions, researchers can circumvent the limitations of natural evolution, creating biocatalysts with enhanced activity, stability, and selectivity. This capability promises to accelerate processes across a broad spectrum of applications, from the efficient synthesis of pharmaceuticals and fine chemicals to the development of sustainable materials and biofuels, ultimately offering solutions to some of the most pressing challenges in chemistry and beyond.
The convergence of generative artificial intelligence and protein engineering presents a transformative opportunity to overcome longstanding hurdles in multiple scientific and industrial fields. Recent studies demonstrate the potential of this technology to dramatically enhance enzyme performance; for example, modifications guided by AI achieved an 80-fold increase in activity for the tP4H enzyme with stabilized variants, alongside a 6-fold improvement in the wild-type version. This level of catalytic enhancement promises significant advancements in areas like drug discovery, where more efficient enzymes can accelerate the synthesis of complex molecules, and sustainable chemistry, offering greener alternatives to traditional chemical processes. Furthermore, the ability to design enzymes with tailored properties extends to materials science, enabling the creation of novel biomaterials and environmentally friendly manufacturing techniques.
The future of enzyme design is rapidly accelerating through innovations in artificial intelligence and validation methodologies. Recent studies demonstrate that AI-driven approaches are not simply optimizing existing enzymes, but are now capable of generating entirely new biocatalysts – enzymes created de novo with tailored functions. This progress is evidenced by a 7.7-fold enhancement in ketosteroid isomerase activity achieved through these techniques, all while maintaining sufficient structural novelty – a minimum sequence identity of just 35% to known enzymes – ensuring these are genuinely new biological entities. This ability to create enzymes largely independent of existing biological blueprints promises to revolutionize fields requiring highly specific catalysts, bypassing the limitations of naturally occurring enzymes and opening doors to previously unattainable chemical transformations.
The pursuit of highly efficient catalysts consistently returns to the principle of transition state stabilization. This design strategy centers on the idea that enzymes accelerate reactions not by altering the overall energy landscape, but by specifically lowering the activation energy – the energetic hurdle reactants must overcome. By engineering enzymes to more closely resemble the high-energy transition state of a reaction, catalytic efficiency is dramatically increased; effectively reducing the energy required for the reaction to proceed. This is achieved through careful manipulation of the enzyme’s active site, introducing interactions that preferentially bind and stabilize the fleeting, unstable intermediate configuration. Recent successes demonstrate this approach remains vital, with engineered enzymes exhibiting substantial activity gains – a testament to the enduring power of understanding and mimicking the energetic preferences of the transition state itself in the ongoing quest for optimized biocatalysts.
The pursuit of novel enzymes, as detailed in this review of generative AI applications, echoes a sentiment articulated by Marie Curie: “Nothing in life is to be feared, it is only to be understood.” The article highlights the iterative process of designing these biocatalysts – repeated model training, failure analysis, and refinement – a process inherently rooted in dismantling the unknown. This isn’t about achieving instant ‘insight’ from colorful dashboards, but rather systematically reducing uncertainty through rigorous testing. The promise of sequence and backbone generators lies not in replacing hypothesis-driven research, but in accelerating the cycle of proposing, testing, and disproving, ultimately moving closer to a genuine understanding of protein structure and function. The more models tested, the better the chance of identifying viable candidates – a principle of relentless experimentation that Curie herself would undoubtedly appreciate.
What’s Next?
The proliferation of generative models in enzyme design represents, predictably, a shift in optimization strategies, not a transcendence of them. A model, after all, is merely a compromise between knowledge and convenience. Current architectures excel at interpolating from existing protein space – a skilled mimicry, certainly – but genuine innovation demands extrapolation. The field now faces the unglamorous task of defining “novelty” itself, and distinguishing between statistically improbable sequences and genuinely functional enzymes. Claims of ‘optimal’ designs should be met with immediate scrutiny; optimal for whom, and under what rigorously defined conditions?
The immediate future likely involves a move beyond sequence generation towards more holistic design. Backbone flexibility, post-translational modifications, and the protein’s microenvironment are routinely simplified, treated as negligible noise. A truly predictive model will necessitate incorporating these complexities, and accepting the corresponding increase in computational cost. More fundamentally, progress hinges on better data. The current reliance on solved structures and characterized enzymes creates a circularity; models learn to reproduce what is already known, reinforcing existing biases.
Ultimately, the true test lies not in the elegance of the algorithm, but in the mundane reality of industrial biocatalysis. Can these in silico enzymes compete with the rugged, imperfect, and often serendipitous products of directed evolution? The answer, one suspects, will reveal less about the power of artificial intelligence and more about the enduring limitations of predictive power itself.
Original article: https://arxiv.org/pdf/2602.03779.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-04 09:12