Author: Denis Avetisyan
A new multi-agent AI framework is streamlining the process of polymer research, from predicting material properties to generating novel designs.
This review details an autonomous system leveraging physics-informed neural networks for high-throughput analysis of both synthetic and biopolymers, incorporating uncertainty quantification and scalable generative design capabilities.
Despite the increasing demand for novel materials, traditional polymer discovery remains a slow and resource-intensive process. This work, ‘Autonomous Multi-Agent AI for High-Throughput Polymer Informatics: From Property Prediction to Generative Design Across Synthetic and Bio-Polymers’, introduces a multi-agent AI framework capable of accelerating materials research by unifying property prediction, generative design, and biopolymer analysis. Demonstrating strong predictive accuracy-reaching [latex]R^2 = 0.89[/latex] for glass-transition temperature-the system also scales efficiently to 10,000 polymers while minimizing computational cost. Could this approach usher in a new era of rapid, AI-driven polymer innovation, enabling the design of materials with tailored properties for diverse applications?
Decoding Polymer Complexity: The Limits of Prediction
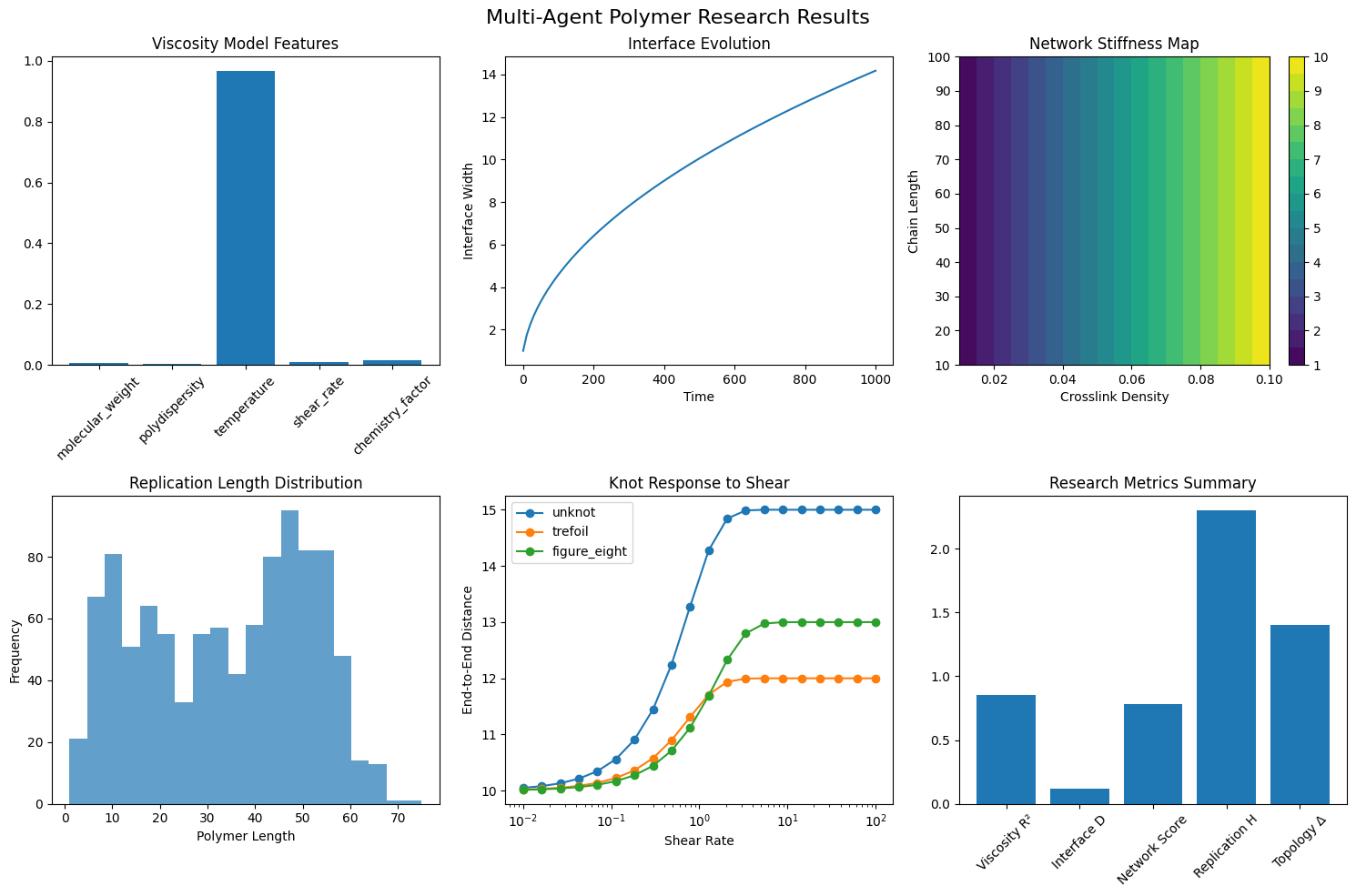
The advancement of materials discovery is fundamentally bottlenecked by the laborious process of characterizing polymer properties. Traditionally, researchers rely on extensive synthesis and physical testing – a cycle that demands significant time, resources, and material expenditure. This conventional approach struggles to keep pace with the vast chemical space of possible polymers, hindering the identification of novel materials with desired characteristics. Consequently, the ability to accurately predict these properties – such as mechanical strength, thermal stability, and solubility – before physical creation is paramount. Computational methods offer a potential solution, but often require substantial processing power and complex simulations to account for the intricate relationship between a polymer’s molecular structure and its macroscopic behavior, creating a continuing need for more efficient predictive tools.
Predicting the macroscopic properties of polymers – characteristics like tensile strength, elasticity, and density – presents a significant challenge due to the inherent complexity of their structures. Unlike simpler materials with repeating, uniform arrangements, polymers exhibit a vast range of molecular weights, branching patterns, and conformational freedom. These structural nuances at the nanoscale directly influence how polymer chains interact and organize, ultimately dictating the material’s bulk behavior. Traditional methods often rely on empirical correlations or computationally expensive simulations that struggle to capture this structural diversity and its cascading effect on macroscopic properties. The intricate relationship between a polymer’s molecular architecture and its resulting performance necessitates advanced modeling techniques capable of bridging the gap between nanoscale structure and observable, real-world characteristics.
Orchestrated Intelligence: A Multi-Agent Approach to Materials Design
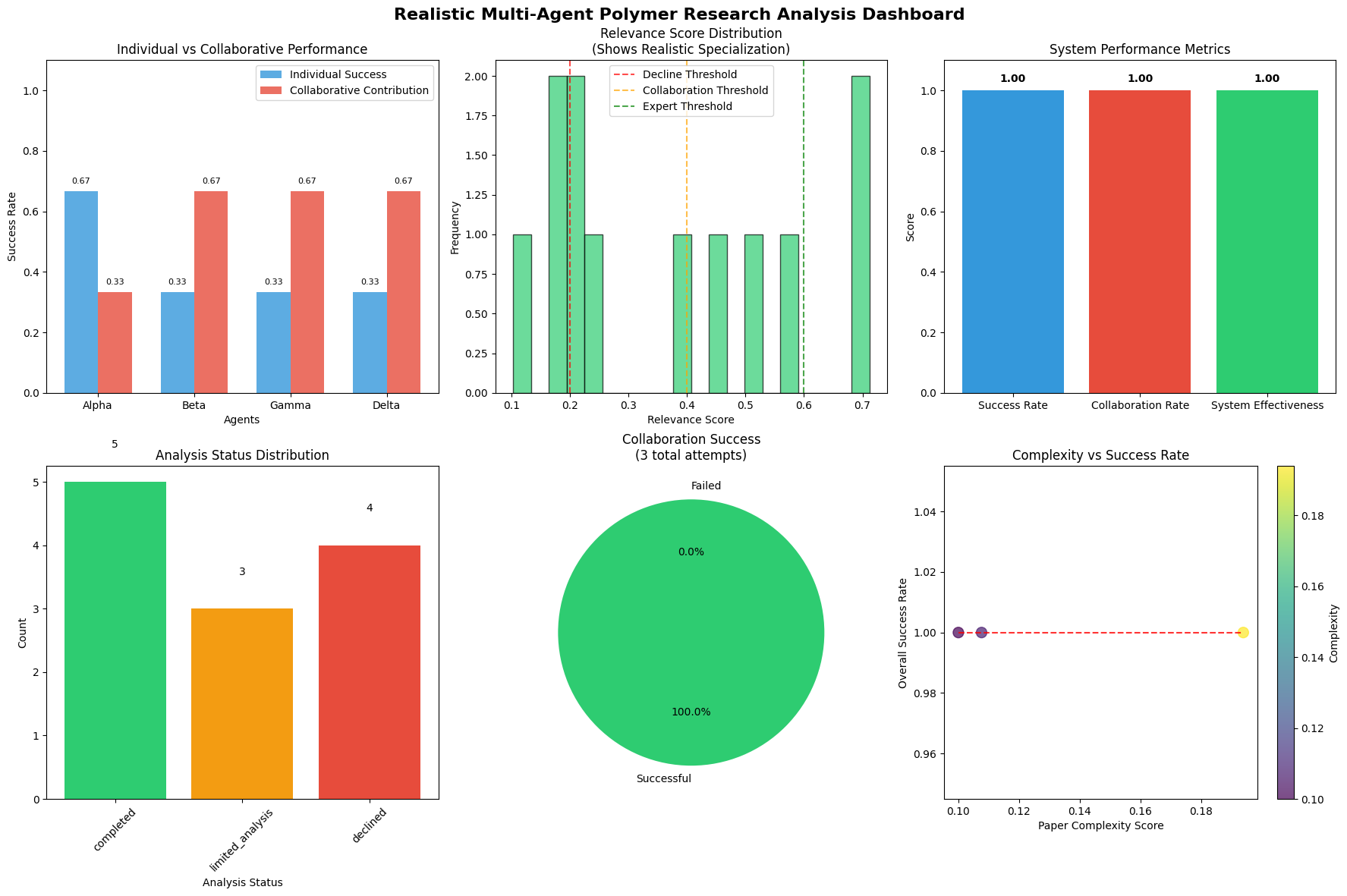
This Multi-Agent System (MAS) is designed to expedite the process of predicting polymer properties and optimizing materials design through distributed computation. The system functions by decomposing the complex task of materials discovery into smaller, manageable sub-tasks handled by specialized software agents. This approach enables parallel processing, significantly reducing the time required for property prediction and materials exploration compared to traditional single-model workflows. The modular nature of the MAS also facilitates continuous integration of new algorithms and data sources, allowing for ongoing refinement of predictive capabilities and adaptation to evolving research needs.
The multi-agent system incorporates specialized agents to maintain data quality and promote interoperability. A key component is the Validation Agent, which filters generated molecular structures represented in SMILES notation, achieving a validity rate of 0.95. This high validity ensures that only chemically reasonable candidate molecules are advanced for property prediction and materials design. Additionally, a Knowledge Graph Agent facilitates the sharing and reuse of chemical knowledge, linking molecular structures with associated properties and experimental data to improve the accuracy and efficiency of the system.
The system’s modular architecture enables parallel processing by distributing computational tasks across independent agents, significantly reducing prediction times for polymer properties. This design also facilitates continuous refinement of predictive models through iterative feedback loops and the incorporation of new data. Agents can be updated and retrained independently without disrupting the overall system functionality, allowing for ongoing improvements in accuracy and efficiency. The ability to dynamically adapt models to new information ensures the system remains current with the latest materials science data and optimizes predictive performance over time.
PolyGNN: Reverse-Engineering Material Behavior with Graph Neural Networks
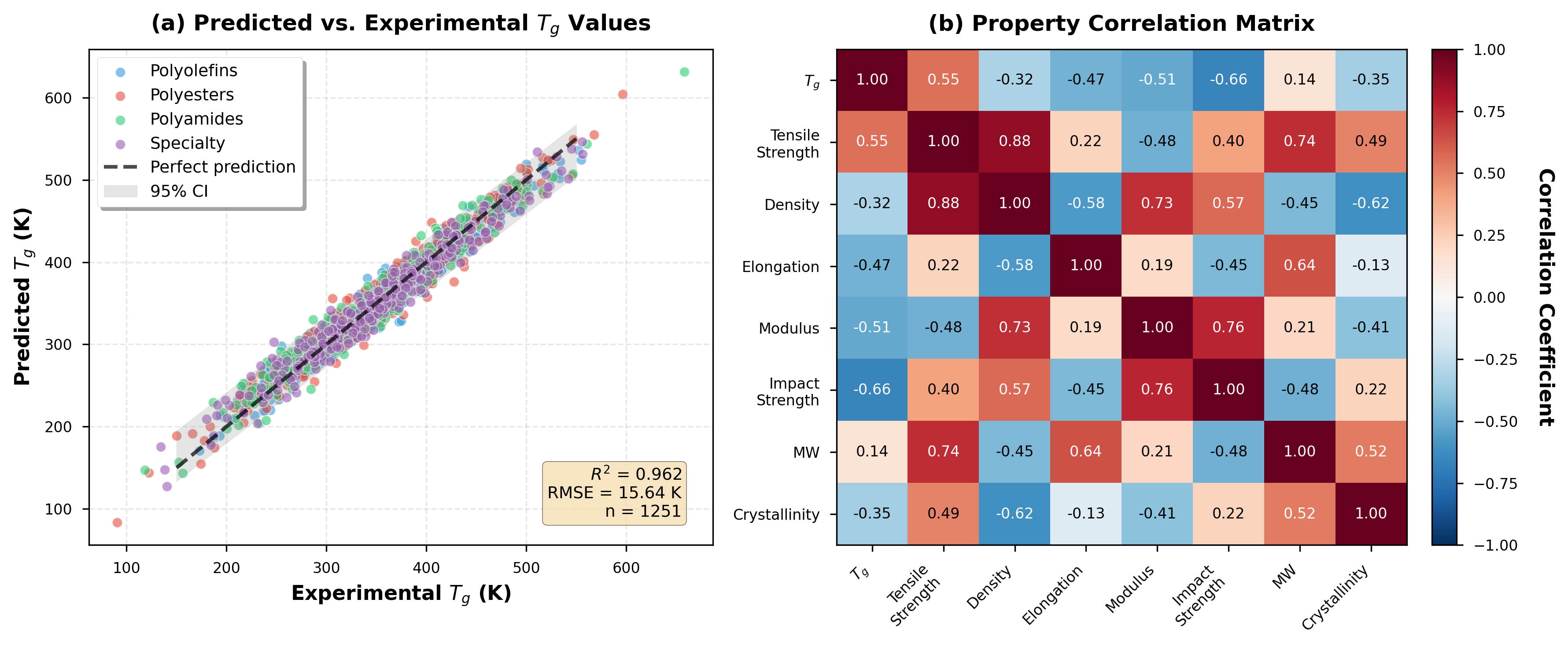
PolyGNN is a graph neural network specifically developed for the prediction of material properties in polymers. The model is trained exclusively on data contained within the PolyInfo Database, a curated collection of polymer structures and their associated properties. By representing polymers as graphs – where atoms are nodes and bonds are edges – PolyGNN can learn relationships between molecular structure and macroscopic properties such as glass transition temperature, tensile strength, and density. This approach allows for quantitative property prediction directly from a polymer’s chemical graph, enabling efficient materials discovery and design.
PolyGNN represents polymers as graphs, where atoms are nodes and chemical bonds are edges, enabling the model to directly process the connectivity information inherent in molecular structure. This graph-based representation allows PolyGNN to learn relationships between a polymer’s topology – branching, crosslinking, and chain length – and its macroscopic properties. Specifically, the model correlates graph features, such as node degree, shortest path lengths, and subgraph patterns, with properties including glass transition temperature (Tg), tensile strength, and density. This approach circumvents the need for manually engineered features and allows the model to automatically discover relevant structural motifs predictive of material behavior.
The PolyGNN model achieves a held-out test set R2 value of approximately 0.98, indicating a high degree of correlation between predicted and actual polymer properties. To further improve predictive performance and model stability, an ensemble method is employed. This technique combines the predictions generated by multiple independently trained PolyGNN instances; the aggregated output reduces variance and increases the robustness of the overall property prediction, leading to more reliable results across diverse polymer structures.
Beyond Prediction: Modeling the Inevitable with Physics-Informed Neural Networks
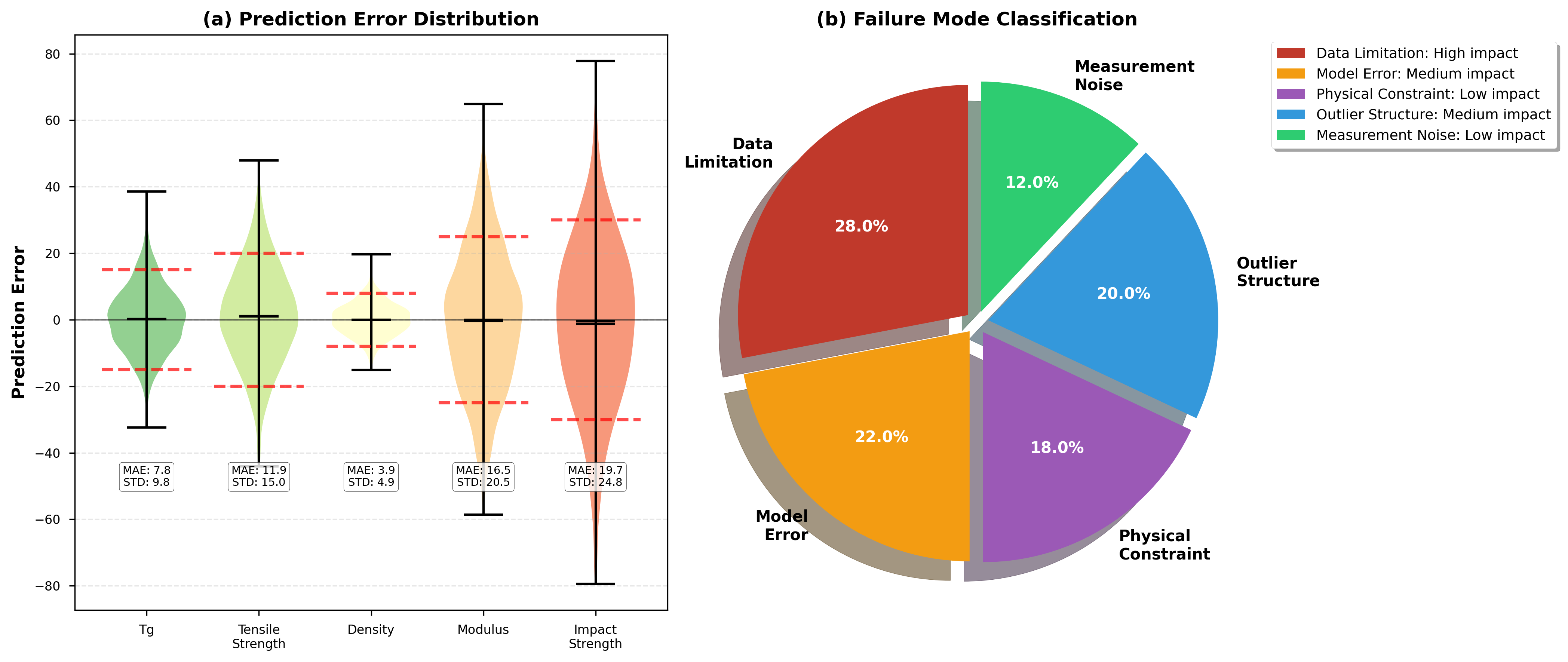
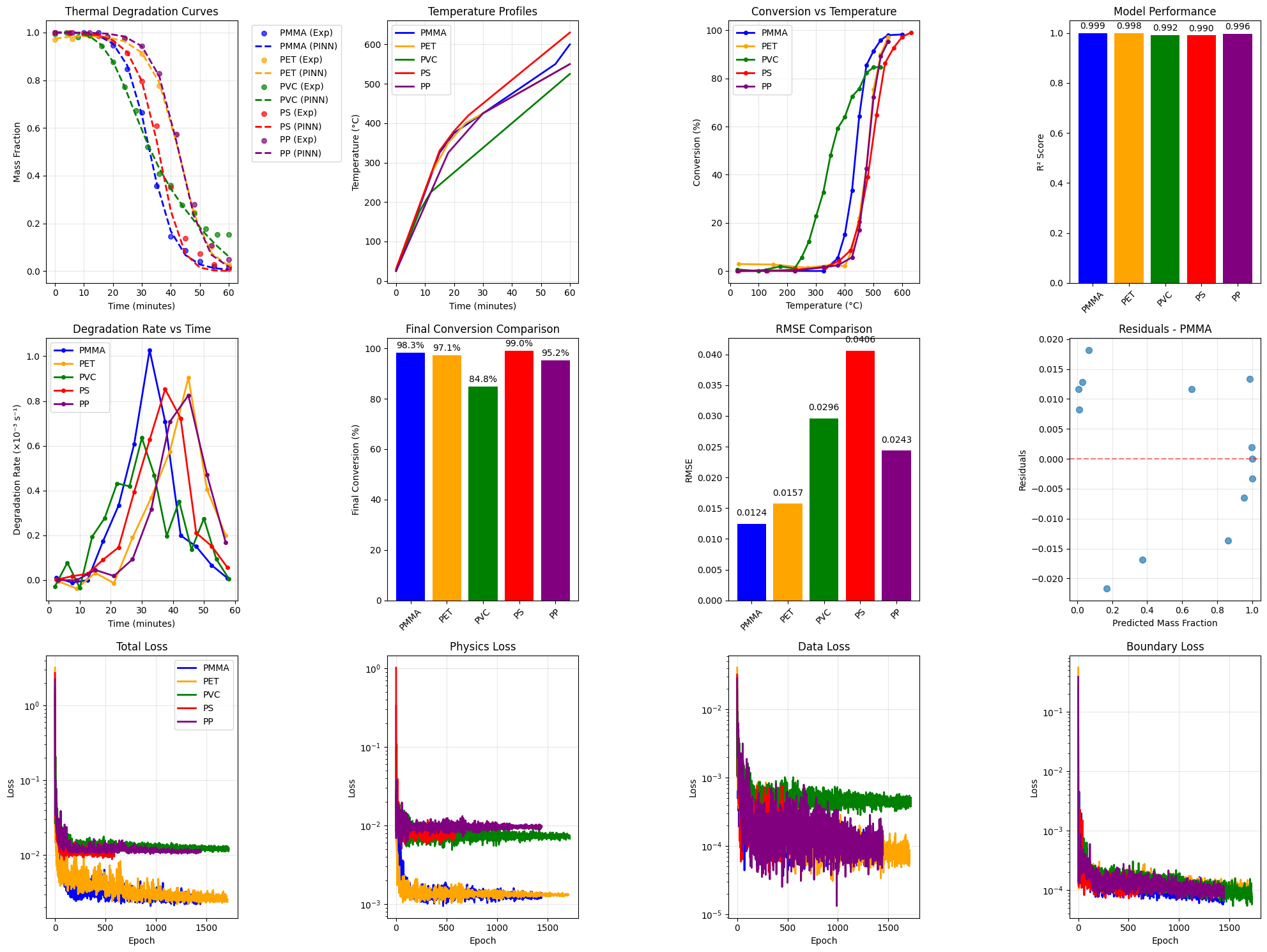
The complex issue of polymer degradation is tackled through the innovative integration of Physics-Informed Neural Networks (PINN) within a multi-agent system. This approach moves beyond traditional data-driven modeling by embedding known physical laws – governing the material’s breakdown – directly into the neural network’s learning process. Rather than solely relying on observed data, the PINN framework allows the system to extrapolate and predict degradation behavior even when faced with conditions outside the initial training dataset. This is achieved by formulating the degradation process as a partial differential equation, which is then used as a constraint during the network’s training. Consequently, the system doesn’t simply learn degradation; it understands it through the lens of established physical principles, leading to more robust and reliable predictions of material lifespan and performance.
Physics-Informed Neural Networks (PINN) offer a powerful approach to modeling polymer degradation by embedding known physical laws directly into the learning process. Unlike traditional neural networks which are purely data-driven, PINN utilizes governing equations – such as diffusion models or kinetic rate laws – as a regularization term during training. This integration ensures the model’s predictions adhere to established physical principles, even when extrapolating beyond the range of available training data. Consequently, PINN can accurately predict polymer behavior under novel conditions or over extended timescales, a crucial capability for assessing material durability and lifespan. By effectively combining the strengths of data-driven learning and physics-based modeling, PINN circumvents the limitations of purely empirical approaches and enables robust, generalizable predictions of polymer degradation phenomena.
The ability to accurately forecast the longevity of polymer materials represents a significant advancement across numerous fields. Traditional methods often struggle to extrapolate beyond the conditions used during testing, limiting predictions of performance under novel stresses or over extended timescales. However, by integrating physics-informed neural networks, researchers can now model polymer degradation processes with greater fidelity, effectively predicting how materials will behave far beyond the scope of initial observations. This capability is particularly crucial for applications demanding high reliability and long service life, such as aerospace components, biomedical implants, and durable infrastructure, where premature failure carries substantial economic and safety risks. Consequently, these predictive models allow for optimized material selection, improved design strategies, and proactive maintenance schedules, ultimately extending the useful life and reducing the lifecycle costs of polymer-based products.
Expanding the Horizon: A Future of Materials Intelligence
The developed materials intelligence framework isn’t limited by its initial demonstration; its architecture is designed for broad applicability across diverse materials classes. This extensibility stems from a modular design, allowing researchers to integrate data from various sources – encompassing inorganic compounds, ceramics, composites, and beyond – into a cohesive system. By standardizing data representation and leveraging shared machine learning algorithms, the framework facilitates the creation of a truly unified platform for materials discovery and design. Such a platform promises to overcome the traditional siloed approach to materials science, enabling predictive modeling and accelerated innovation across a wide spectrum of technological challenges, from energy storage and conversion to advanced manufacturing and sustainable materials development.
The current materials intelligence framework possesses significant potential for expansion into the realm of biomaterials and complex polymer-protein conjugates through synergistic integration with advanced protein structure prediction tools like AlphaFold2. By leveraging AlphaFold2’s capacity to accurately determine protein three-dimensional structures from amino acid sequences, the system can move beyond traditional materials to design and optimize novel biocompatible materials. This integration would enable the prediction of how proteins interact with synthetic polymers at a molecular level, facilitating the creation of materials with tailored properties for applications in drug delivery, tissue engineering, and biosensing. The resulting platform would not only predict material characteristics but also offer insights into the protein-polymer interface, paving the way for a new generation of intelligently designed biomaterials with enhanced functionality and performance.
The system’s computational efficiency is demonstrated through rapid analysis times – an average of 3.2 minutes for protein analysis and just 30 seconds to predict the characteristics of five synthetic polymers. This speed facilitates high-throughput materials exploration, suggesting a pathway towards accelerated innovation. Researchers anticipate that future iterations will leverage AI-powered multi-agent systems to further streamline the discovery process, potentially tackling complex challenges in areas such as sustainable material design and the development of advanced technologies, ultimately enabling a more responsive and efficient materials development cycle.
![A UMAP projection of polymer chemical space demonstrates distinct clustering by family and smooth gradients of [latex]T_g[/latex] and synthetic accessibility, with star markers highlighting successfully validated novel polymer designs.](https://arxiv.org/html/2602.00103v1/Images/UMAP.png)
The framework detailed in this study, with its emphasis on distributed intelligence through multi-agent systems, operates as a deliberate stress test of conventional polymer informatics. It’s a system designed to expose limitations – to find where predictive models falter and generative designs break down. This approach aligns with Jürgen Habermas’s assertion: “The world speaks to us through the medium of labor.” Here, the ‘labor’ is the AI’s iterative process of prediction and design, constantly revealing the inherent constraints and ‘design sins’ within the data and algorithms. By pushing the boundaries of uncertainty quantification and integrating diverse data, the system doesn’t merely predict; it actively diagnoses the weaknesses of the existing knowledge base, much like reverse-engineering a complex material’s properties.
Uncharted Territory
The presented framework, while demonstrating a clear advance in automated polymer informatics, merely scratches the surface of what’s possible. It operates, fundamentally, as a sophisticated pattern-matching engine. The true challenge isn’t simply predicting properties-any sufficiently complex system can be trained to do that-but understanding why those properties emerge. Reality, after all, is open source-it’s just that the code is written in a language far older and more intricate than anything currently available to a neural network. The next iteration must move beyond correlation and begin to genuinely reverse-engineer the underlying physics and chemistry.
A significant limitation lies in the reliance on existing datasets. While the system capably integrates diverse data types, it remains constrained by the biases and gaps inherent in those sources. The real leap forward will necessitate a shift towards active learning-systems that intelligently design experiments to fill knowledge gaps and validate their own hypotheses. Furthermore, the current focus on property prediction and generative design overlooks the crucial element of processability. A perfectly designed polymer is useless if it cannot be synthesized or manufactured at scale.
Ultimately, the pursuit of AI-driven materials discovery is not about replacing human intuition, but about augmenting it. The framework offers a powerful tool for navigating the vast chemical space of polymers, but the interpretation of results, the formulation of new questions, and the recognition of truly novel materials will still require the uniquely human capacity for creative insight. The code is complex, but the compiler still needs a skilled programmer.
Original article: https://arxiv.org/pdf/2602.00103.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-03 19:52