Author: Denis Avetisyan
A new model accurately decodes biological activity from tissue images, offering a powerful way to analyze cancer pathways without relying on traditional genomic data.
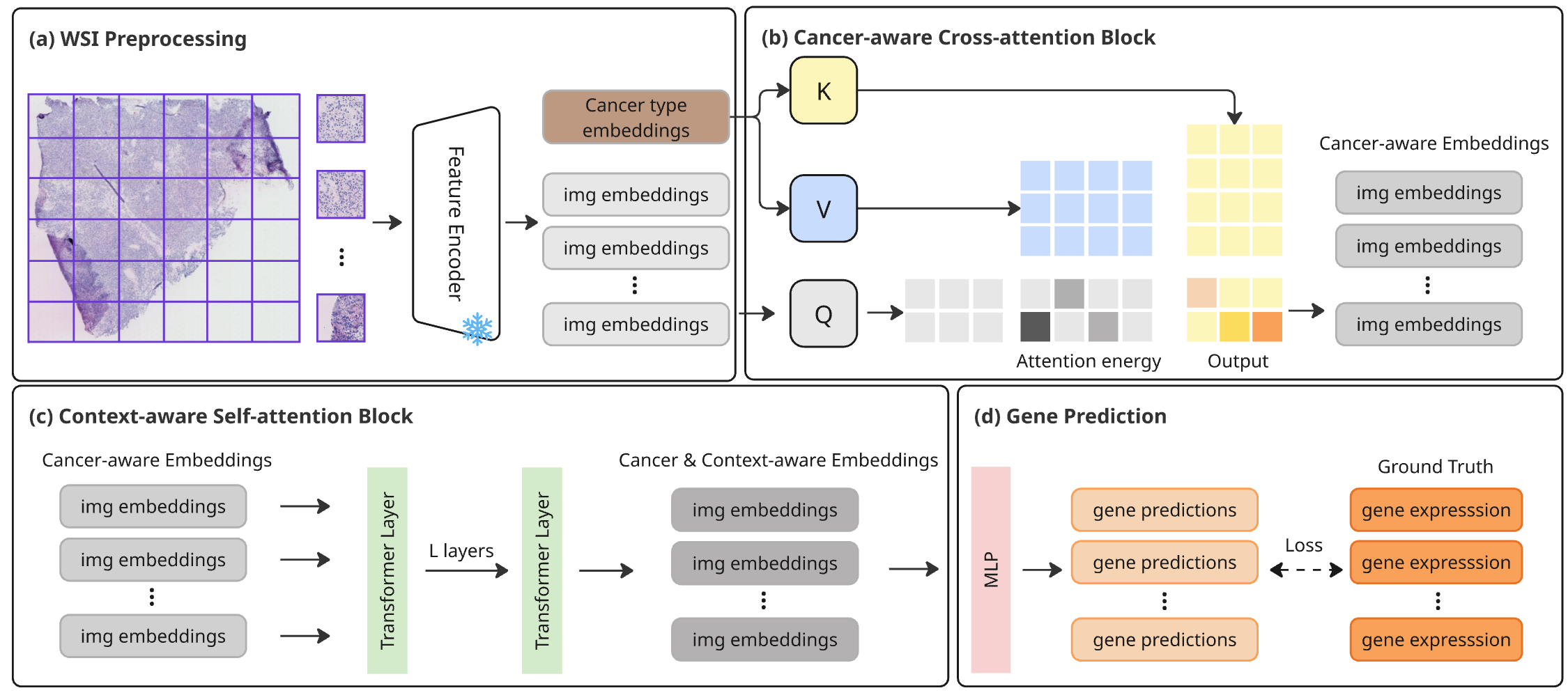
HistoPrism, a transformer-based approach, predicts gene expression from pan-cancer histology images and introduces a pathway coherence metric for improved biological evaluation.
While spatially resolved transcriptomics offers powerful insights into cancer biology, its clinical translation is hindered by cost and accessibility. To address this, we present ‘HistoPrism: Unlocking Functional Pathway Analysis from Pan-Cancer Histology via Gene Expression Prediction’, a transformer-based model that accurately predicts gene expression directly from routinely available H&E-stained histology images across multiple cancer types. Critically, HistoPrism not only surpasses existing methods in predicting individual gene expression, but also demonstrates improved performance at the level of biologically coherent functional pathways. Does this pathway-level accuracy represent a key step towards realizing the clinical potential of computationally derived transcriptomic information from whole slide images?
The Erosion of Context: Morphology and the Genomic Blindspot
Historically, genomic studies have largely focused on identifying genetic mutations and alterations, often proceeding without significant consideration of the tissue’s observable characteristics – its morphology. This separation represents a critical gap in understanding disease mechanisms, particularly in complex illnesses like cancer. While genomic data can reveal what is changing at a molecular level, morphological analysis – examining the size, shape, and organization of cells and tissues – provides crucial context about where and how these changes manifest. Ignoring morphology means potentially overlooking vital clues about disease progression, treatment response, and the interplay between genetic drivers and the tumor microenvironment. Consequently, a purely genomic approach can lead to incomplete interpretations and missed opportunities for developing targeted therapies that address both the genetic and the visible hallmarks of disease.
Histopathology images, particularly those utilizing hematoxylin and eosin (H&E) staining, present a significant analytical hurdle despite being a cornerstone of cancer diagnosis. The inherent complexity arises from the sheer volume of data contained within a single image – encompassing cellular morphology, tissue architecture, and staining intensity – resulting in extremely high dimensionality. This vastness often overwhelms traditional image analysis techniques, making it difficult to discern subtle yet critical biological signals indicative of disease state or treatment response. Effectively extracting meaningful features requires navigating a complex interplay of variables and overcoming challenges related to image variability, staining artifacts, and the subjective nature of morphological assessment, ultimately demanding sophisticated computational approaches to bridge the gap between visual observation and quantifiable biological insight.
Cancer research stands to gain significantly from analytical approaches that synthesize visual information derived from histopathology – the study of tissues – with comprehensive genomic data. Currently, these data types are often analyzed in isolation, potentially obscuring critical relationships between a tumor’s genetic makeup and its observable characteristics at the cellular level. Integrating these modalities allows for a more nuanced understanding of disease progression, treatment response, and the identification of novel biomarkers. Such integrative methods promise to move beyond simple genetic correlations and reveal how genomic alterations manifest morphologically, ultimately leading to more precise diagnostics and personalized therapeutic strategies. This holistic view is crucial, as the genetic blueprint of a cancer doesn’t fully explain its behavior; the way those genes are expressed and alter cellular structure, as seen under the microscope, provides an equally vital piece of the puzzle.
The Rise of Foundation Models: A Shift in Pathology’s Paradigm
Pathology Foundation Models (PFMs), including UNI PFM and Gigapath PFM, are developed through training on exceptionally large datasets of histopathology images. This large-scale pre-training allows the models to learn generalized visual representations, capturing fundamental patterns and features present within tissue morphology. The resulting models do not require task-specific labeling for initial feature extraction; instead, they can be adapted to a variety of downstream tasks with minimal fine-tuning. The robustness of these learned representations stems from the diversity and scale of the training data, enabling PFMs to perform effectively even with limited labeled data for specific applications.
Pre-training pathology foundation models (PFMs) on large-scale histopathology image datasets allows them to learn generalized visual features independent of specific diagnostic tasks. This process involves exposing the model to millions of images, enabling it to identify patterns and structures relevant to cellular morphology and tissue organization. Consequently, these learned representations can be transferred and fine-tuned for various downstream applications, including gene expression prediction, where the model learns to correlate visual features with underlying molecular data. The benefit is a reduction in the need for task-specific labeled data, as the pre-trained model already possesses a strong understanding of relevant image characteristics, improving performance and efficiency in subsequent analyses.
STPath is a pan-cancer foundation model designed to predict gene expression directly from histopathology images. Utilizing a masked gene modeling approach, the model is trained to reconstruct missing gene expression values based on visual features extracted from tissue samples. This allows STPath to establish a correlation between the morphological characteristics observed in images and the underlying genomic activity of cells, effectively bridging the gap between visual pathology and molecular biology. The model’s pan-cancer scope indicates its training encompassed data from multiple cancer types, enabling generalization across different malignancies and potentially facilitating the prediction of gene expression in novel or rare cancers.
Beyond Correlation: Assessing Biological Fidelity with Gene Pathway Coherence
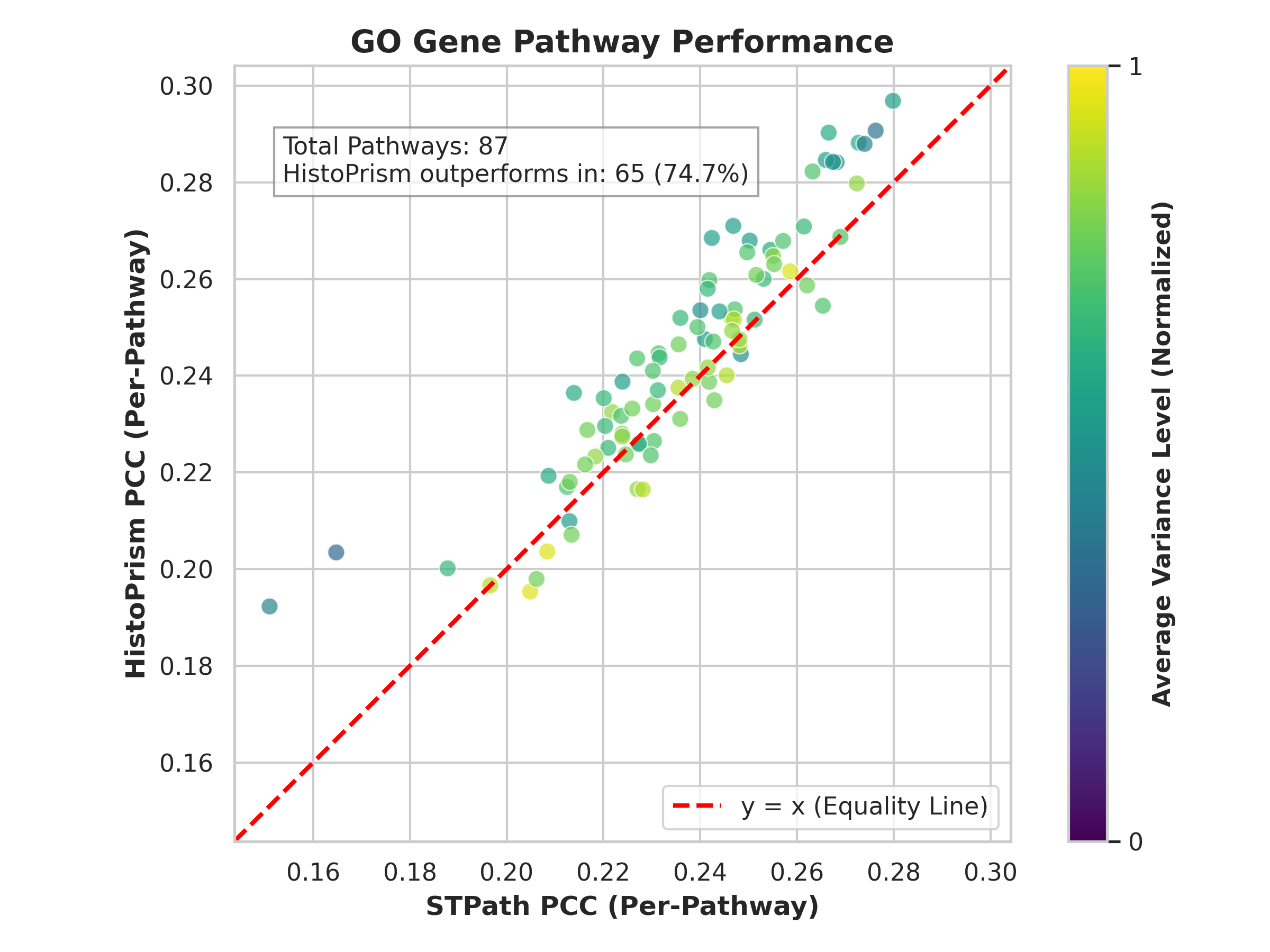
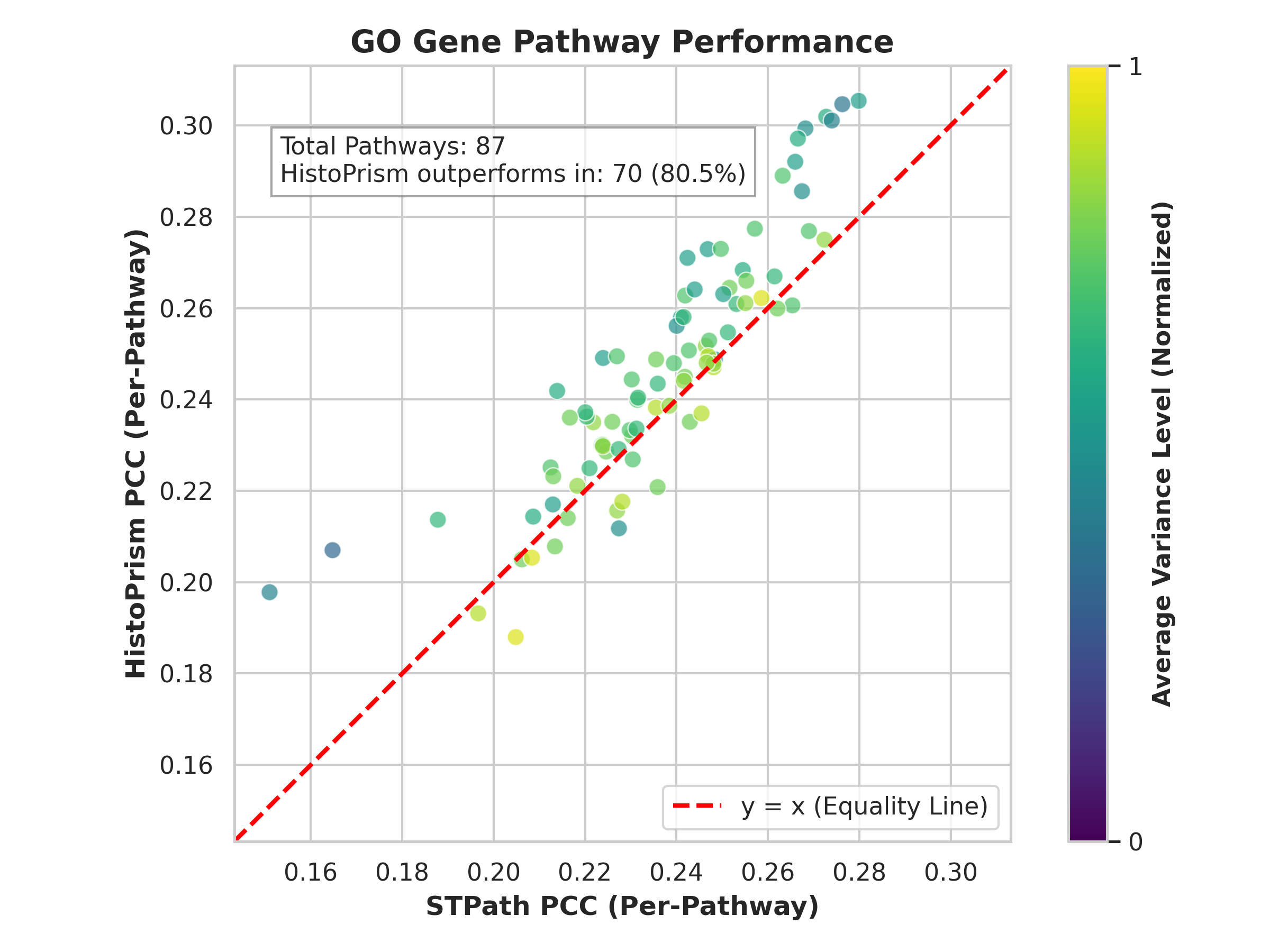
Gene Pathway Coherence (GPC) represents a departure from traditional methods of validating predicted gene expression which often rely on simple correlation with known ground truth data. GPC assesses the alignment of predicted expression patterns with established biological processes by evaluating their coherence with curated gene sets, specifically Gene Ontology (GO) pathways and Hallmark gene sets. This approach moves beyond assessing individual gene prediction accuracy to determine if the predicted changes in gene expression collectively reflect meaningful alterations in known biological pathways. By focusing on pathway-level agreement, GPC offers a more robust and biologically relevant metric for evaluating the fidelity of predicted gene expression data, providing insights into whether the predictions represent genuine biological signals rather than spurious correlations.
Gene Pathway Coherence (GPC) assesses the degree to which predicted gene expression changes correspond to established biological knowledge, specifically through analysis of Gene Ontology (GO) Pathways and Hallmark Gene Sets. This is accomplished by evaluating whether predicted gene expression patterns exhibit statistically significant correlation with genes known to participate in these predefined biological processes. Rather than simply measuring correlation with individual genes, GPC determines if the overall predicted expression profile reflects the coordinated activity expected within functionally related gene groups. A high GPC score indicates that the predicted expression changes are not random, but rather align with known biological mechanisms and pathways, providing a more biologically relevant validation metric than methods focused solely on individual gene prediction accuracy.
Gene Pathway Coherence (GPC) quantitatively assesses the alignment of predicted gene expression with established biological pathways by employing Pearson correlation calculations focused on highly variable genes. This approach moves beyond single-gene assessments to evaluate the collective behavior of gene sets known to participate in specific biological processes. Highly variable genes, those exhibiting the greatest expression fluctuations, are prioritized in this correlation analysis as they contribute most significantly to pathway-level changes. The resulting Pearson correlation coefficient provides a numerical metric of pathway coherence, with higher values indicating stronger agreement between predicted and expected gene expression patterns within that pathway. This methodology offers a robust and statistically grounded means of evaluating the biological relevance of predictive models, providing a more nuanced assessment of prediction quality than simple correlation with ground truth data.
HistoPrism’s predictive capabilities were validated using the Gene Pathway Coherence (GPC) framework, resulting in a measured coherence of 86.0% when assessed against Hallmark gene pathways and 74.7% coherence with Gene Ontology pathways. This performance demonstrates a substantial improvement over the STPath method, indicating that HistoPrism’s predicted gene expression patterns align more strongly with established biological processes as defined by these curated pathway sets. These GPC scores quantitatively support the biological relevance and fidelity of HistoPrism’s predictions compared to the baseline STPath approach.
Adjusted Mutual Information (AMI) was employed to quantitatively compare the global clustering results of HistoPrism with those of STPath. AMI, a metric for assessing the similarity between two clusterings, yielded substantially higher values for HistoPrism compared to STPath, indicating a greater degree of agreement between HistoPrism’s predicted cell type assignments and established ground truth data. This statistically significant improvement in AMI scores provides independent validation of HistoPrism’s enhanced predictive power and its ability to accurately recapitulate known cellular organization within tissue samples, surpassing the performance of the STPath algorithm in this assessment.
The Pan-Cancer Landscape: A Vision of Integrated Pathology and Precision Oncology
HistoPrism, when integrated with Graph Property Convolution (GPC), offers a powerful framework for comprehensively analyzing cancer across diverse types. This innovative combination allows researchers to not only pinpoint shared genomic and transcriptomic characteristics that underpin multiple cancers, but also to delineate the unique molecular signatures that distinguish each malignancy. By systematically comparing and contrasting these features, HistoPrism reveals fundamental biological mechanisms common to cancer development, alongside those that drive specific tumor behaviors. This pan-cancer approach promises to move beyond a disease-centric view of cancer, fostering a more holistic understanding of its underlying principles and ultimately accelerating the development of broadly effective therapies.
HistoPrism leverages the power of cross-attention mechanisms within its Transformer architecture to effectively merge visual data from histopathology images with complex genomic information. This innovative approach allows the model to identify subtle, yet critical, relationships between a tumor’s appearance and its underlying genetic makeup. By attending to relevant features in both modalities simultaneously, HistoPrism surpasses traditional methods that treat these data types in isolation. Consequently, the model achieves improved predictive accuracy in tasks such as cancer subtype classification and prognosis, offering a more holistic and nuanced understanding of tumor characteristics and potentially leading to more informed clinical decisions.
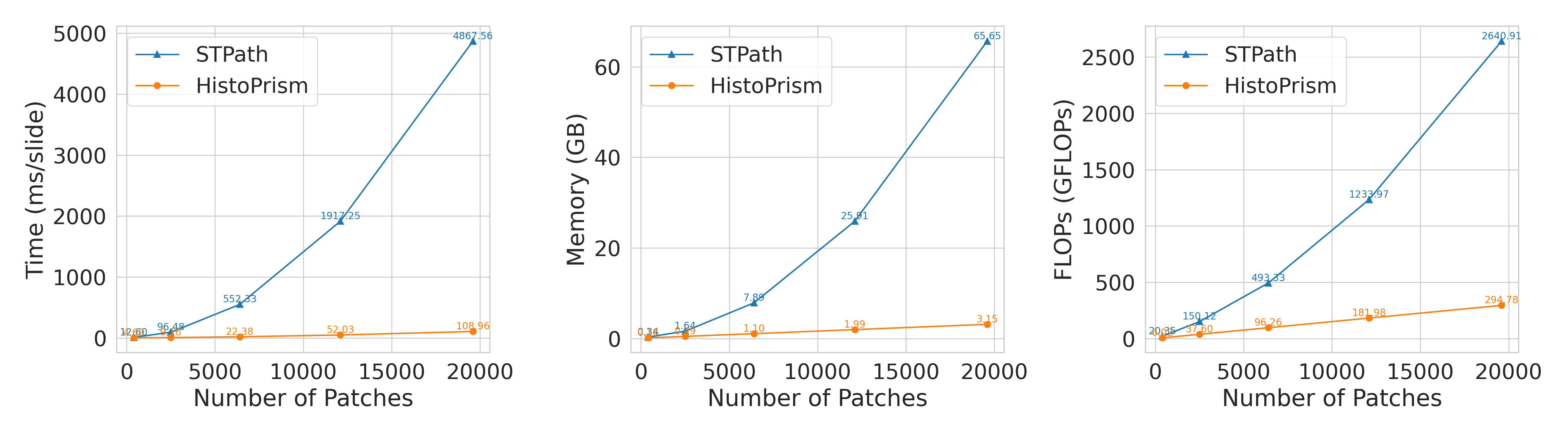
HistoPrism demonstrates a significant leap in computational efficiency when compared to the STPath methodology, addressing a crucial bottleneck in large-scale pathology image analysis. Evaluations reveal that HistoPrism requires substantially less peak GPU memory and fewer floating point operations – or FLOPs – to achieve comparable, and often superior, results. This reduction in computational demand isn’t merely incremental; it allows for the analysis of larger datasets and more complex models without necessitating prohibitively expensive hardware. Critically, HistoPrism’s efficiency scales linearly with increasing data volume, a stark contrast to STPath’s exponential growth in computational requirements, suggesting that HistoPrism will remain viable as datasets continue to expand and become increasingly detailed.
A significant advancement offered by HistoPrism lies in its computational scalability. Unlike existing methods, such as STPath, which experience an exponential increase in processing demands as data volume grows, HistoPrism maintains a linear relationship between data size and computational effort. This means that doubling the amount of input data only requires a proportional increase in processing power, making large-scale, pan-cancer analyses far more feasible. This efficient scaling is crucial for unlocking insights from the ever-increasing availability of multi-omics data and ultimately accelerating the development of more effective, personalized cancer treatments by enabling analyses previously constrained by computational limitations.
The integration of computational pathology and genomic analysis, as demonstrated by advancements in tools like HistoPrism, is poised to revolutionize cancer treatment by enabling a truly personalized approach. Historically, treatment decisions have often been guided by broad categorizations of cancer type and stage; however, this research suggests a future where therapies are meticulously crafted to match the individual characteristics of each patient’s tumor. By comprehensively analyzing both the visual morphology of cancer cells and their underlying genomic signatures, clinicians can move beyond generalized protocols and identify subtle yet critical distinctions that dictate treatment response. This refined understanding promises to optimize therapeutic efficacy, minimize adverse effects, and ultimately improve outcomes for individuals facing a cancer diagnosis, moving the field closer to precision oncology.
Ongoing research aims to integrate Spatial Transcriptomics data with existing HistoPrism analyses, promising a more nuanced understanding of tumor biology. This next step will move beyond traditional genomic and morphological assessments by mapping gene expression directly onto the spatial organization of cells within the tumor microenvironment. By revealing how gene activity varies across different regions of the tumor, and how these variations relate to cell-cell interactions and immune infiltration, researchers anticipate significantly refined predictive models. This detailed spatial resolution has the potential to identify previously hidden therapeutic targets and biomarkers, ultimately enabling more precise and effective personalized cancer treatments tailored to the unique characteristics of each patient’s tumor ecosystem.
The pursuit of predictive power, as demonstrated by HistoPrism’s gene expression forecasting from histological images, feels less like construction and more like tending a garden. The model doesn’t build understanding so much as reveal the coherence already present within the biological system. This resonates with a sentiment expressed by Blaise Pascal: “The belly is the window to the soul.” While seemingly disparate, both observations highlight the futility of imposing external structure. HistoPrism doesn’t dictate pathway activity; it reads the existing signatures, much like one infers internal states from external manifestations. Each deployment isn’t a triumph of engineering, but an acknowledgment of inevitable approximation – a prophecy of the limits of prediction itself, even with sophisticated transformer architectures.
What Lies Ahead?
HistoPrism offers a compelling, if predictable, demonstration of predictive power. The model’s success isn’t surprising – histology, after all, is gene expression made visible, albeit through the distorting lens of cellular morphology and staining artifacts. The real challenge isn’t better prediction, but accepting the inevitable divergence between inferred and actual biological activity. Monitoring is the art of fearing consciously; each improved prediction is simply a more refined articulation of what will eventually fail to correlate.
The emphasis on pathway coherence represents a necessary, though imperfect, shift. Biological systems aren’t collections of genes, but negotiated agreements between them. However, coherence metrics remain brittle proxies for true functional integration. The focus should move beyond evaluating what is predicted, towards understanding why predictions degrade – what systemic stresses, what unmodeled interactions, precipitate the inevitable loss of signal? That’s not a bug – it’s a revelation.
True resilience begins where certainty ends. The future isn’t about building more accurate models, but cultivating systems that gracefully accommodate – even require – their own failures. HistoPrism, and its successors, should be viewed not as tools for definitive diagnosis, but as probes for uncovering the inherent instability of biological networks. The question isn’t ‘how well does the image predict the gene?’ but ‘how does the failure of prediction reveal the system’s hidden vulnerabilities?’
Original article: https://arxiv.org/pdf/2601.21560.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-02-01 02:02