Author: Denis Avetisyan
A new machine learning framework accelerates the discovery of promising metal hydrides for efficient and scalable hydrogen storage.
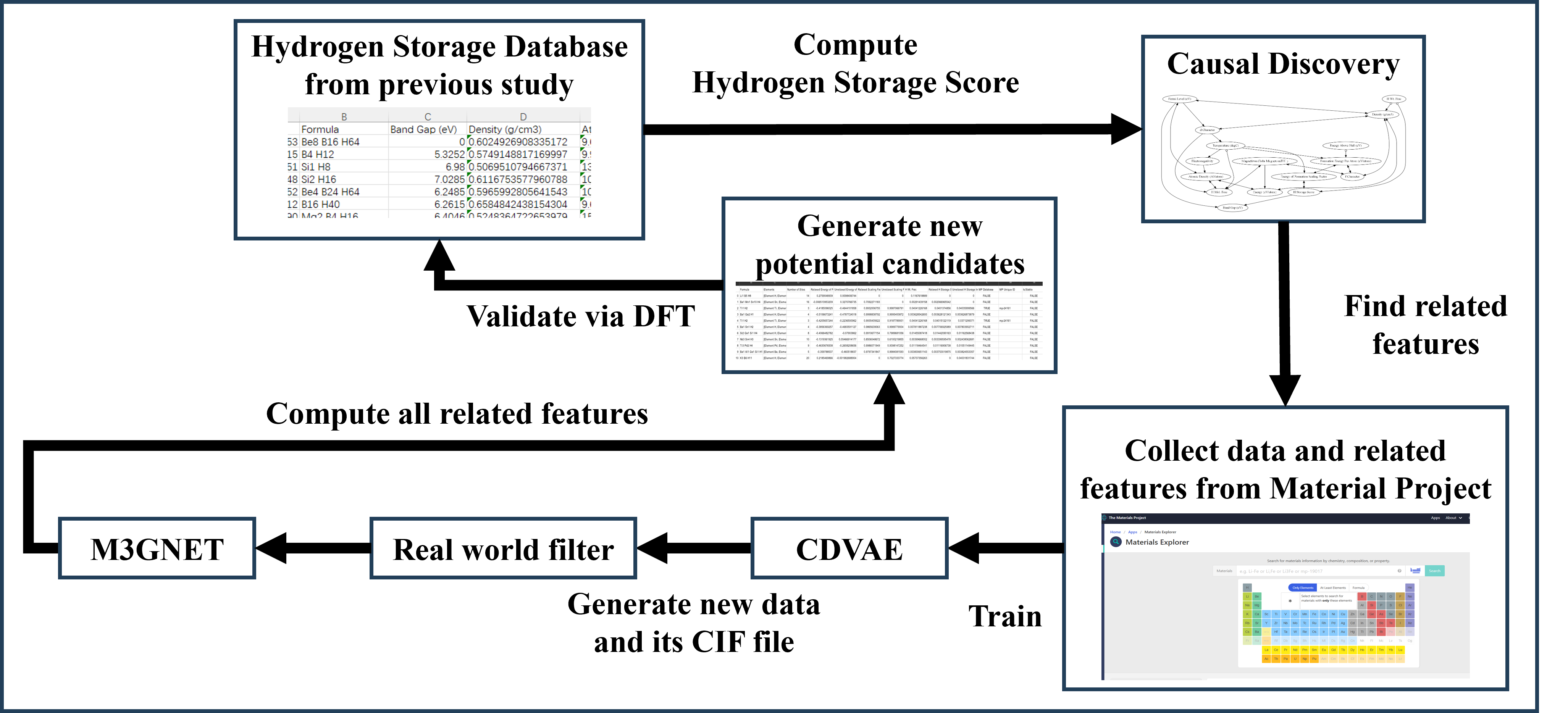
Researchers combine causal discovery and generative modeling to identify and validate four novel metal hydride candidates through DFT simulations.
Despite growing interest in hydrogen as a clean energy carrier, the discovery of efficient and stable hydrogen storage materials remains a significant challenge. This is addressed in ‘A generative machine learning model for designing metal hydrides applied to hydrogen storage’, which presents a novel framework integrating causal discovery with generative machine learning to design previously unknown metal hydride candidates. The approach successfully generated and validated four promising new materials from a dataset of 450 samples, expanding beyond existing materials databases. Could this scalable and time-efficient methodology accelerate the identification of next-generation hydrogen storage solutions and facilitate the transition to carbon-neutral energy systems?
Unlocking the Hydrogen Storage Bottleneck: A Materials Challenge
The promise of a hydrogen economy – a sustainable energy future powered by this abundant element – is currently constrained by significant material science challenges. While hydrogen holds immense potential as a clean fuel, effectively storing it remains a critical bottleneck; current materials simply lack the capacity to hold sufficient hydrogen for practical applications, like powering vehicles or providing grid-scale energy storage. Beyond capacity, the kinetics of hydrogen adsorption and release are also problematic – many materials absorb hydrogen too slowly for rapid refueling or power delivery, and releasing it often requires energy-intensive processes. This combination of low storage density and sluggish reaction rates necessitates the development of entirely new materials capable of both efficiently capturing and readily releasing hydrogen, a pursuit that will determine the feasibility of a widespread transition to this clean energy carrier.
The pursuit of efficient hydrogen storage has long been hampered by the laborious and costly nature of traditional materials discovery. Historically, researchers have relied on synthesizing and testing numerous candidate materials, a process that demands significant resources and time – often years – to identify even a single promising compound. This “trial-and-error” approach isn’t merely slow; the synthesis of each new material requires specialized equipment and skilled personnel, contributing to substantial financial burdens. Furthermore, the vastness of the materials space – the nearly infinite combinations of elements and structures – means that many potentially superior hydrogen storage materials may remain undiscovered simply due to the impracticality of exhaustively exploring all possibilities. Consequently, progress toward viable, large-scale hydrogen storage solutions has been significantly delayed by this inherent bottleneck in materials development.
Predicting how effectively novel materials can store hydrogen remains a significant hurdle due to the inherent complexities of modeling this interaction. Current computational techniques, while powerful, often simplify the intricate interplay of quantum mechanical effects, material defects, and hydrogen’s delicate bonding with surfaces. These simplifications introduce inaccuracies when estimating key performance indicators like storage capacity and kinetics-how quickly hydrogen can be adsorbed and released. Specifically, accurately capturing the many-body effects arising from the collective behavior of electrons and atoms within the material proves computationally demanding, especially for complex materials with varied compositions and structures. Consequently, simulations frequently deviate from experimental results, necessitating costly and time-consuming trial-and-error material synthesis and testing to identify promising candidates for practical hydrogen storage applications.
![Analysis of the Hydrogen Storage Database using the FCI algorithm reveals statistical dependencies-represented by edges, including correlations indicated by bidirectional lines-between the hydrogen storage score and key material features like hydrogen weight fraction ([latex]WH_{2} [/latex]), formation energy ([latex]E_{form} [/latex]), band gap, and crystal structure (f Character) at a significance level of [latex] \alpha=0.05 [/latex].](https://arxiv.org/html/2601.20892v1/FCIresult.png)
Accelerating Discovery: Machine Learning as a Predictive Tool
Machine learning (ML) techniques are increasingly utilized to expedite the identification of novel hydrogen storage materials by establishing correlations between a material’s structural characteristics and its hydrogen storage capacity. This predictive capability bypasses the traditionally slow and resource-intensive process of trial-and-error material synthesis and characterization. ML models are trained on datasets comprising structural features – such as atomic coordinates, crystal structure types, and surface area – paired with experimentally determined or computationally derived hydrogen storage metrics, including gravimetric and volumetric densities, adsorption enthalpies, and kinetics. Successful models allow researchers to screen large databases of potential materials in silico, prioritizing those with predicted performance characteristics favorable for hydrogen storage applications, thereby reducing experimental workloads and accelerating materials discovery.
Classical Machine Learning (ML) techniques, such as Grand Canonical Monte Carlo (GCMC) and Reverse Monte Carlo (RMC), historically served as initial screening methods in materials discovery due to their established frameworks for property prediction. GCMC simulations, for example, assess the thermodynamic stability of materials at varying conditions, while RMC refines structural models to match experimental data. However, the efficacy of these methods is significantly constrained by their reliance on substantial datasets of material properties and structural information for both training and validation. Generating these datasets through experiment or computationally expensive ab initio calculations presents a bottleneck, limiting the scope of materials that can be effectively screened and hindering the exploration of wider chemical spaces. Consequently, while providing a foundational approach, classical ML methods often require complementary techniques to overcome their data limitations.
Deep learning methodologies, particularly Graph Neural Networks (GNNs), demonstrate enhanced predictive capabilities for materials properties compared to classical machine learning techniques. However, the performance of these models is strongly correlated with the size and quality of the training dataset. GNNs require substantial data to accurately learn the complex relationships between a material’s atomic-level structure and its macroscopic properties; insufficient data leads to overfitting and poor generalization to unseen materials. Data requirements stem from the large number of parameters within deep neural networks, necessitating extensive examples to constrain the model and avoid spurious correlations, and ensuring reliable prediction of hydrogen storage capacity and related metrics.
Designing Materials from First Principles: Generative Modeling Emerges
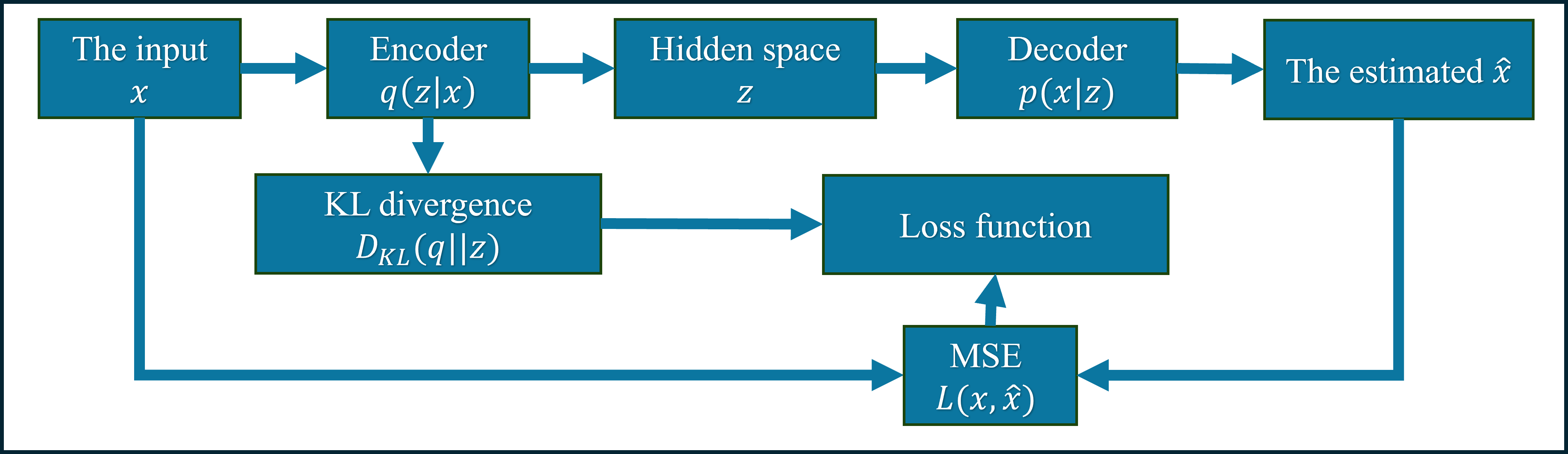
Variational Autoencoders (VAEs) represent a class of generative models capable of learning the complex, multi-dimensional structure inherent in datasets of known hydrogen storage materials. These models function by encoding existing material structures into a lower-dimensional latent space, effectively capturing the essential features defining their properties. Once trained, the VAE can sample from this latent space to generate novel material structures that statistically resemble the training data. By incorporating specific constraints or objectives during the sampling process – such as desired hydrogen storage capacity or stability – researchers can tailor the generated materials to exhibit pre-defined, optimized characteristics. This in silico approach accelerates material discovery by proposing candidates with a higher probability of possessing the targeted properties, reducing the need for extensive and costly trial-and-error experimentation.
Crystal Diffusion Variational Autoencoders (CDVAE) represent a recent advancement in materials discovery, utilizing Diffusion Probabilistic Models to generate viable crystal structures. These models operate by progressively adding noise to training crystal structures until they become pure noise, then learning to reverse this process – denoising from random noise to create new, structurally valid configurations. Unlike traditional generative models, CDVAE excels at generating diverse and realistic structures by modeling the probability distribution of crystal structures directly. The diffusion process, combined with the variational autoencoder framework, enables the generation of structures that adhere to crystallographic principles and possess desired characteristics, offering a powerful tool for in silico materials design.
The performance of Crystal Diffusion Variational Autoencoders (CDVAE) in materials design is directly dependent on the accuracy of property prediction for generated structures. This is commonly achieved by integrating CDVAE with Density Functional Theory (DFT) calculations, which provide first-principles estimates of material properties. However, DFT calculations can be computationally expensive; therefore, machine learning models like M3GNet are frequently used to accelerate the structure relaxation process and provide a rapid initial assessment of stability and properties before more precise DFT calculations are performed. The combination of CDVAE with DFT and M3GNet enables both the generation of viable candidate materials and efficient evaluation of their performance characteristics, such as hydrogen storage capacity and binding energy.
Fast Causal Inference (FCI) methods are employed to determine the relationship between specific structural features of hydrogen storage materials and their resulting Hydrogen Storage Score (HSS). These techniques move beyond simple correlation analysis by identifying causal links – which features directly influence the HSS, rather than merely appearing associated with it. By applying FCI, researchers can pinpoint key structural determinants, such as pore size, surface area, or specific atomic arrangements, that have a demonstrable impact on hydrogen adsorption and storage capacity. This knowledge is then integrated into the generative modeling process, allowing algorithms to prioritize the creation of materials exhibiting these identified features, thereby increasing the probability of generating high-performing hydrogen storage candidates. The process leverages observational data and statistical inference to build a causal graph representing these relationships, enabling targeted material design.
Beyond Prediction: A Paradigm Shift in Materials Discovery
The search for novel materials with tailored properties is often hampered by the sheer size of the possible compositional space. Recent advances leverage the power of generative modeling, coupled with machine learning-driven property prediction, to navigate this complexity with unprecedented efficiency. This approach doesn’t simply analyze existing materials; it proactively creates potential candidates, effectively charting a course through the vast chemical landscape of alloy and metal hydrides. By learning the underlying principles governing material stability and performance from a limited dataset, the framework can generate entirely new structures with a high probability of possessing desired characteristics, dramatically accelerating the materials discovery process and reducing reliance on costly trial-and-error experimentation. This computational strategy promises to unlock a new era of materials design, enabling the rapid identification of compounds optimized for specific applications, such as efficient hydrogen storage.
A novel machine learning framework demonstrated the potential for accelerated materials discovery by successfully generating four previously unknown alloy hydride candidates suitable for hydrogen storage. Remarkably, this generative model achieved this feat while trained on a limited dataset of only 270 samples – a significant reduction compared to conventional models requiring tens of thousands of structures. The generated candidates were not simply theoretical constructs; their viability was confirmed through rigorous Density Functional Theory (DFT) simulations, establishing a pathway for efficient in silico screening. This approach drastically reduces the reliance on time-consuming and expensive trial-and-error experimentation, offering a promising strategy for designing next-generation hydrogen storage materials with tailored properties and improved performance.
The predictive power of this new generative model is particularly striking when considering the limited training data utilized – a mere 270 samples. Remarkably, the model achieved a Mean Squared Error (MSE) of 0.064 when predicting alloy hydride formation energy. This level of accuracy is exceptionally close to that of the widely-respected M3GNet model, which required a dataset of 188,000 structures to achieve an MSE of 0.0754. This suggests the framework efficiently learns underlying chemical principles, allowing for robust predictions despite the small training set and highlighting its potential to accelerate materials discovery where data is scarce.
The predictive power of this new generative framework is underscored by its remarkably low Mean Absolute Error (MAE) of 0.0775 when forecasting material formation energy. This level of accuracy is particularly noteworthy considering the model was trained on a dataset of only 270 samples – a fraction of the 188,000 structures used to train the established M3GNet model. The close correspondence in MAE values – 0.0775 for this work versus 0.0775 for M3GNet trained on the extensive Materials Project database – demonstrates the efficiency of this approach, highlighting its potential to accelerate materials discovery with limited data and computational resources. This indicates the generative model effectively captures the underlying principles governing alloy hydride stability, achieving comparable predictive capability despite a significantly smaller training set.
The conventional process of materials discovery is often hampered by extensive trial-and-error, requiring significant resources and years of research to identify promising candidates. However, a recent advance in computational materials science demonstrates the potential to accelerate this process dramatically. By leveraging generative machine learning models, researchers can now virtually synthesize and screen countless material compositions at a fraction of the traditional cost and time. This computational approach not only streamlines the search for novel materials but also allows for the targeted design of materials with specific, desired properties – a capability previously limited by the sheer scale of the chemical space. The rapid generation and in silico evaluation of alloy hydrides, as demonstrated with a model trained on a surprisingly small dataset, represents a significant step toward a future where materials discovery is driven by intelligent algorithms rather than purely empirical methods.
“`html
The research detailed in this study embodies a spirit of intellectual dismantling. It doesn’t simply accept established limitations in material science, but actively seeks to deconstruct and rebuild understanding of hydrogen storage. This pursuit aligns perfectly with Brian Kernighan’s observation: “Debugging is twice as hard as writing the code in the first place. Therefore, if you write the code as cleverly as possible, you are, by definition, not smart enough to debug it.” The team approached the design of metal hydrides not through incremental improvements, but by challenging fundamental assumptions and generating entirely new candidates. By employing a framework combining causal discovery and generative models, the researchers effectively ‘debugged’ the existing landscape of material possibilities, revealing four previously unknown compounds-a testament to the power of questioning established norms and reverse-engineering reality to achieve innovation.
Unlocking the Source Code
The presented work doesn’t simply offer new hydrogen storage materials; it demonstrates a methodology. Reality, after all, is open source – the underlying rules exist, but are obscured by complexity. This framework, blending causal discovery with generative modeling, represents a step toward reverse-engineering those rules, at least within the specific domain of metal hydrides. The successful generation and validation of novel candidates suggest the approach isn’t merely stochastic, but guided by a nascent understanding of the compositional space and its relationship to desired properties.
However, the limitations are, predictably, illuminating. The reliance on Density Functional Theory (DFT) simulations, while necessary, introduces inherent approximations. The ‘ground truth’ remains elusive, and the generated materials are only ‘promising’ relative to the model’s predictive power. The true test lies in scalability – can this framework efficiently navigate the vast chemical space, and will the generated candidates consistently translate to practical, real-world performance?
Future work should address these questions by incorporating experimental feedback loops, moving beyond purely computational validation. Further refinement of the causal discovery algorithms could also reveal more fundamental relationships, potentially allowing for the design of materials with properties beyond those currently considered. The ultimate goal isn’t just to find better hydrogen storage materials, but to develop a generalizable methodology for materials discovery-a way to systematically read the code of reality itself.
Original article: https://arxiv.org/pdf/2601.20892.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-01-31 02:33