Author: Denis Avetisyan
Researchers have developed a novel framework that leverages contextual information and causal reasoning to accurately predict how molecules will behave, even with minimal training data.
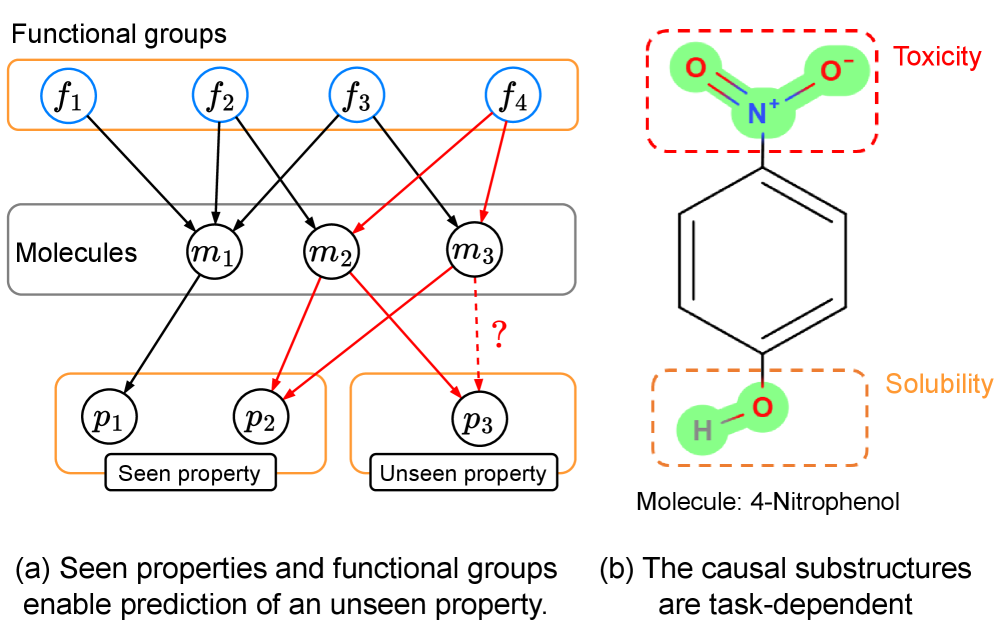
This paper introduces CaMol, a context-aware graph causality inference framework for few-shot molecular property prediction based on identifying key causal substructures and intervention strategies.
Predicting molecular properties is increasingly vital, yet performance severely degrades when labeled data is scarce-a common challenge in drug discovery and materials science. To address this, we present ‘Context-aware Graph Causality Inference for Few-Shot Molecular Property Prediction’, a novel framework, CaMol, that infers causal relationships within molecular structures to improve prediction accuracy with limited examples. CaMol leverages chemical knowledge and a learnable intervention strategy to identify key substructures directly linked to desired properties, achieving superior performance and interpretability. Could this approach unlock more efficient and reliable molecular design through enhanced understanding of structure-property relationships?
The Data Scarcity Challenge in Molecular Discovery
The advancement of both drug discovery and materials science is fundamentally reliant on the accurate prediction of molecular properties, yet this process is hampered by a significant need for vast datasets. Determining characteristics like a molecule’s stability, reactivity, or binding affinity traditionally demands extensive experimentation, which is both time-consuming and expensive. Each property requires numerous examples of molecules with known characteristics to train predictive models; without these large, labeled datasets, the development of new pharmaceuticals or high-performance materials is considerably slowed. Consequently, the pursuit of novel compounds is often bottlenecked not by theoretical understanding, but by the practical limitations of data acquisition, necessitating innovative approaches to overcome this critical challenge and accelerate scientific progress.
Conventional machine learning algorithms, while powerful, often demand vast quantities of meticulously labeled data to accurately predict molecular properties – a significant obstacle in fields like drug discovery and materials science. The creation of these datasets is expensive, time-consuming, and frequently limited by the sheer number of possible molecular configurations. Consequently, models trained on insufficient data exhibit poor generalization, struggling to reliably predict the characteristics of novel compounds. This data scarcity effectively bottlenecks innovation, as researchers find it difficult to computationally screen potential candidates and accelerate the identification of promising materials or therapeutic interventions. The reliance on large datasets thus presents a fundamental challenge, prompting a search for alternative learning strategies that can overcome these limitations and unlock the full potential of computational molecular discovery.
The intricate three-dimensional arrangements of atoms within molecules present a significant hurdle for conventional machine learning approaches. Unlike images or text, where patterns can emerge from relatively small datasets, molecular properties are deeply influenced by subtle structural variations-a slight change in bond angle or conformation can drastically alter a molecule’s behavior. This sensitivity demands vastly larger datasets to achieve reliable predictions, but acquiring such data is often expensive, time-consuming, or even impossible. Consequently, researchers are increasingly focused on data-efficient learning paradigms-techniques like transfer learning, meta-learning, and active learning-that can extract maximal information from limited datasets and generalize effectively to unseen molecular structures, promising a pathway towards accelerated discovery in both chemistry and materials science.
Meta-Learning: A Strategy for Learning with Limited Data
Meta-learning addresses the challenge of limited data in molecular property prediction by training models to learn how to learn. Traditional machine learning requires substantial datasets for each new property prediction task; meta-learning, conversely, aims to develop models capable of quickly generalizing to new tasks with only a small number of examples – a capability termed ‘few-shot learning’. This is achieved by exposing the model to a distribution of related tasks during training, enabling it to learn a prior or initialization that facilitates rapid adaptation when faced with a novel molecular property prediction problem. The core principle is to optimize not for performance on a single task, but for the ability to learn any task efficiently, thereby overcoming the data scarcity common in drug discovery and materials science.
Model-Agnostic Meta-Learning (MAML) and Prototype Networks (ProtoNet) achieve efficient knowledge transfer by learning a shared representation space across multiple related tasks. MAML optimizes for initial model parameters that can be quickly fine-tuned with a small number of gradient steps on a new task, effectively learning an initialization that is sensitive and adaptable. ProtoNet, conversely, learns a metric space where examples from different classes cluster around prototype representations calculated from support examples; new examples are classified by their proximity to these prototypes. Both methods rely on the principle of transferring learned information – either in the form of optimized parameters or a learned distance metric – to accelerate learning on unseen, but related, tasks, thereby reducing the need for extensive task-specific training data.
Direct application of meta-learning algorithms, such as MAML and ProtoNet, to molecular graphs presents challenges due to the inherent complexity of representing molecular structure. Unlike images or sequences, molecular graphs are non-Euclidean and require specialized feature encoding methods to capture relevant chemical information. Standard graph neural networks (GNNs) are typically employed, but careful consideration must be given to the choice of GNN architecture, node and edge features, and message passing schemes to ensure effective learning of transferable representations. Furthermore, the variable size and connectivity of molecular graphs necessitate techniques to handle differing graph topologies and avoid issues with fixed-size input requirements of some meta-learning algorithms. Efficiently capturing long-range interactions and conformational flexibility within the graph structure also remains a critical consideration for robust meta-learning performance.
CaMol: A Framework for Causal Molecular Property Prediction
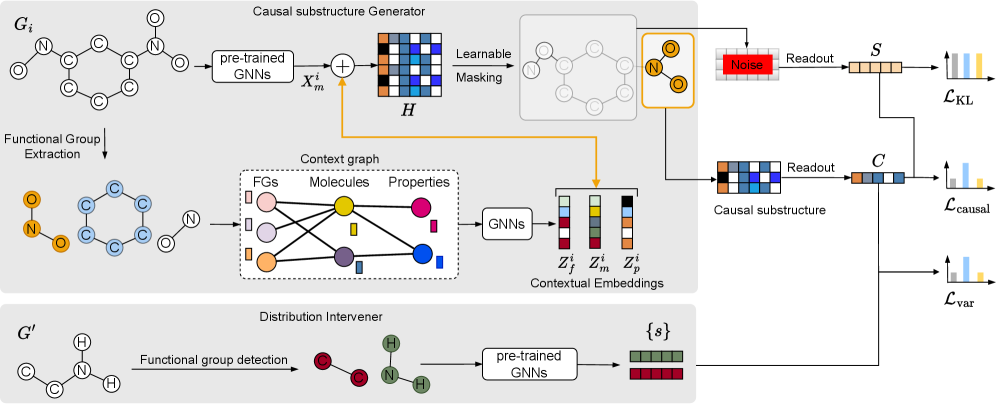
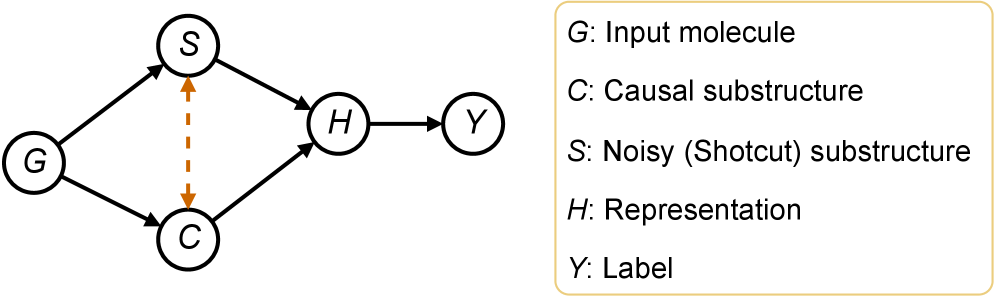
CaMol is a new framework designed for predicting molecular properties when limited training data is available – a scenario known as few-shot learning. The system integrates graph neural networks (GNNs) to process molecular structures represented as graphs, with causal inference techniques to determine the relationship between substructures and predicted properties. This approach moves beyond simple correlation-based predictions by attempting to model the underlying causal mechanisms, enabling more accurate and generalizable predictions even with sparse data. The framework aims to identify which molecular features are causally responsible for specific property values, rather than simply identifying features that are statistically associated with those values.
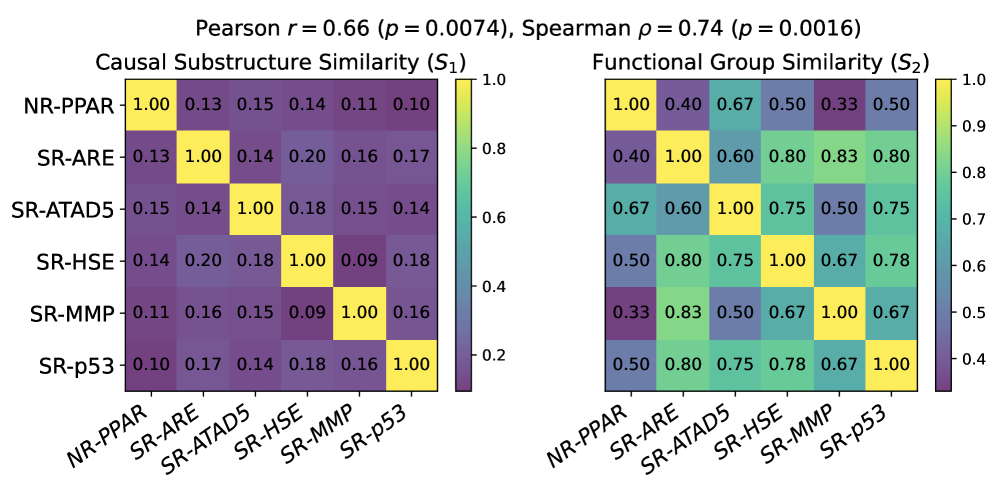
CaMol utilizes a ‘Context Graph’ to represent the relationships between molecular functional groups and target properties, enabling identification of causal substructures. This graph explicitly defines how specific functional groups contribute to, or influence, predicted properties, moving beyond simple correlative feature extraction. The Context Graph is constructed by analyzing the contribution of each functional group to the overall molecular property prediction, allowing the model to determine which substructures are causally linked to the target value. This explicit modeling facilitates more accurate predictions, particularly in few-shot learning scenarios where data is limited, as it prioritizes understanding the underlying causal mechanisms rather than relying on statistical associations.
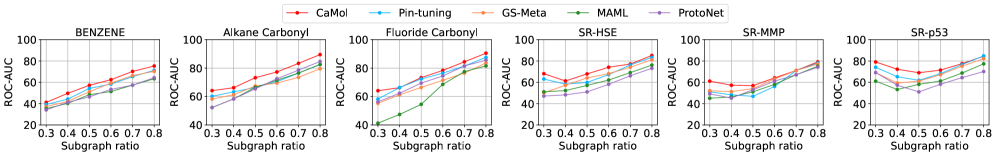
CaMol utilizes node masking in conjunction with the pre-trained S-CGIB molecular encoder to improve focus on salient molecular features during property prediction. Node masking randomly deactivates a percentage of nodes within the molecular graph, forcing the model to rely on the remaining, potentially more informative substructures. This, combined with the representational power of S-CGIB – which provides a strong initial molecular encoding – results in enhanced performance. Quantitative evaluation demonstrates CaMol achieves state-of-the-art results, exhibiting an average relative improvement of 7.36% across multiple benchmark datasets compared to existing methods.
Unlocking Molecular Understanding Through Causal Inference
CaMol distinguishes itself from typical predictive models by moving beyond simply what a molecule will do to revealing why it behaves in a certain manner. The model doesn’t just forecast properties; it dissects molecular structures to pinpoint the substructures – specific arrangements of atoms – that are most influential in driving those properties. This is achieved through a novel framework that assesses the contribution of each substructure, effectively highlighting the crucial chemical features responsible for observed behaviors. Consequently, researchers gain a deeper understanding of the structure-activity relationship, allowing for a more rational and targeted approach to molecular design and optimization, rather than relying solely on empirical observations or trial-and-error methods.
CaMol advances beyond simple molecular property prediction by actively seeking to understand the reasons behind those properties, achieved through the implementation of causal inference techniques. The model employs methods like backdoor adjustment – statistically controlling for confounding variables that might falsely link a substructure to a specific behavior – and intervention, which simulates the effect of altering a specific molecular feature. This allows CaMol to distinguish correlation from causation; for example, determining if a substructure truly causes increased toxicity, or merely appears associated with it due to other underlying factors. By disentangling these complex relationships, researchers gain a more reliable basis for designing molecules with desired characteristics, moving beyond trial-and-error towards a more rational and efficient approach to discovery.
The ability to not only predict, but also explain molecular behavior significantly streamlines both drug discovery and materials design processes. Recent advancements, exemplified by the CaMol model, prioritize compounds with a higher probability of success by revealing the specific substructures driving desired properties. This interpretability allows researchers to move beyond simple screening, focusing efforts on molecules with demonstrably favorable characteristics. Validating this approach, CaMol achieved a robust performance of 86.89% on the Tox21 dataset – a significant improvement over existing methods, exceeding their average performance by 7.36%. This enhanced predictive power, coupled with causal insights, promises to substantially accelerate the identification of promising candidates and reduce the time and resources required to bring new innovations to fruition.
Future Directions: Expanding the Scope of Causal Molecular Learning
Continued development of CaMol prioritizes expanding its capabilities to handle increasingly large and intricate molecular datasets, a crucial step towards real-world applicability. Current research focuses not only on computational efficiency to process these expanded datasets, but also on enhancing the model’s robustness against noisy or incomplete data – a common challenge in chemical experimentation. Improving generalizability is also key; the goal is for CaMol to accurately infer causal relationships not just within the specific datasets used for training, but across diverse molecular structures and chemical properties. Successfully addressing these challenges will unlock CaMol’s potential as a broadly applicable tool for accelerating drug discovery, materials science, and other fields reliant on understanding molecular mechanisms.
The convergence of causal molecular learning, as exemplified by CaMol, with generative modeling presents a compelling pathway towards de novo molecular design. By leveraging CaMol’s ability to discern true causal relationships from observational data, generative models can move beyond simply mimicking known structures and instead focus on creating molecules optimized for specific, desired properties. This integration allows for the systematic exploration of chemical space, potentially yielding compounds with enhanced efficacy, selectivity, or stability – attributes difficult to achieve through traditional trial-and-error methods. The resulting AI systems promise not only to accelerate drug discovery but also to unlock innovative materials with tailored functionalities, moving beyond incremental improvements to truly novel chemical entities.
Advancing the theoretical underpinnings of causal molecular learning promises to unlock the potential for increasingly sophisticated and transparent artificial intelligence systems in drug discovery and materials science. Recent work, exemplified by the CaMol framework, demonstrates the practical benefits of this approach; on the BENZENE dataset, CaMol achieved a substantial reduction in distributional divergence, specifically a 36.6% decrease in Jensen-Shannon Divergence (JSD) when contrasted with the leading baseline model and a 32.3% improvement over the next best performing method. These results not only highlight CaMol’s efficacy in discerning complex molecular relationships, but also suggest that a deeper theoretical understanding of causality within molecular systems will be crucial for building AI capable of designing and optimizing novel compounds with desired characteristics, moving beyond correlation to genuine understanding.
The pursuit of efficient molecular property prediction, as detailed in the framework CaMol, mirrors a dedication to distilling complex systems into their essential components. The methodology champions identifying causal substructures – a process akin to removing extraneous detail to reveal underlying mechanisms. This resonates with John McCarthy’s assertion: “It is better to be elegantly concise than to be perfectly verbose.” CaMol’s focus on few-shot learning-achieving accuracy with limited data-demands a similar parsimony. The framework doesn’t attempt to model every interaction; instead, it prioritizes the critical causal pathways, embodying a principle of clarity over exhaustive detail. The elegance of the approach lies in its ability to extract meaningful predictions from sparse information, aligning with McCarthy’s emphasis on effective communication through conciseness.
Where Do We Go From Here?
The pursuit of predictive power with limited data invariably leads to frameworks – they called it CaMol – attempting to distill signal from noise. This work, rightly, focuses on why a molecule exhibits a property, rather than merely that it does. The identification of causal substructures is a laudable goal, though one suspects the true complexity of molecular interactions will continue to exceed any neatly defined ‘context graph.’ The temptation to add layers – attention mechanisms within graph networks, meta-learning algorithms to optimize the few shots – will be strong. Perhaps resisting that urge is the key.
A genuine advance will likely hinge not on more elaborate architectures, but on better definitions of ‘context.’ Current approaches treat it as a static property of the molecule itself. A more nuanced view might consider the experimental conditions, the solvent, even the history of the sample. These are messy details, of course, and thus often ignored. But nature rarely conforms to the convenient boundaries of a dataset.
The field will eventually confront a simple truth: perfect prediction with sparse data is an illusion. The goal, then, shifts from minimizing error to maximizing useful error. Identifying the cases where the model fails – and understanding why – may prove more valuable than achieving incremental gains in overall accuracy. A model that knows its limits is, after all, a far more trustworthy companion.
Original article: https://arxiv.org/pdf/2601.11135.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Clash Royale Best Boss Bandit Champion decks
- Vampire’s Fall 2 redeem codes and how to use them (June 2025)
- World Eternal Online promo codes and how to use them (September 2025)
- Best Arena 9 Decks in Clast Royale
- Country star who vanished from the spotlight 25 years ago resurfaces with viral Jessie James Decker duet
- ‘SNL’ host Finn Wolfhard has a ‘Stranger Things’ reunion and spoofs ‘Heated Rivalry’
- JJK’s Worst Character Already Created 2026’s Most Viral Anime Moment, & McDonald’s Is Cashing In
- Solo Leveling Season 3 release date and details: “It may continue or it may not. Personally, I really hope that it does.”
- Kingdoms of Desire turns the Three Kingdoms era into an idle RPG power fantasy, now globally available
- M7 Pass Event Guide: All you need to know
2026-01-20 08:58