Author: Denis Avetisyan
A new statistical framework analyzes patterns of amino acid relationships to predict protein interactions and understand the impact of genetic changes.
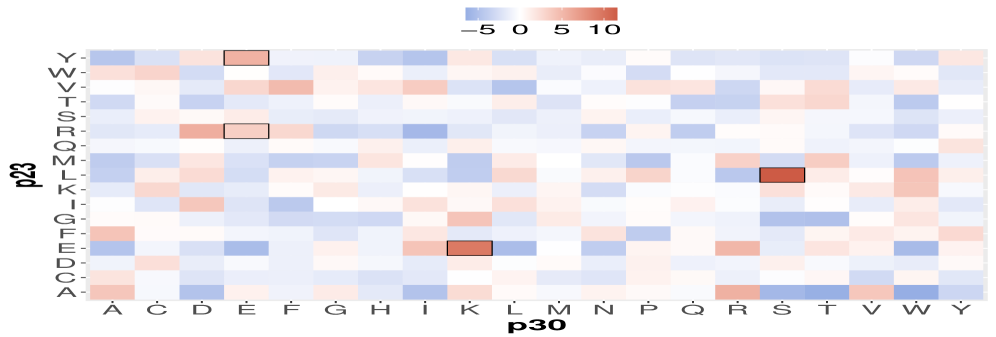
This work introduces CATParc, a model-free approach leveraging partial correlation to characterize protein mutations through coevolutionary analysis and improve residue contact prediction.
While predicting the functional consequences of protein mutations remains a central challenge in molecular biology, existing methods often lack robust statistical inference and model-free approaches. This is addressed in ‘Model-Free Inference for Characterizing Protein Mutations through a Coevolutionary Lens’, which introduces a novel framework-CATParc-leveraging partial correlation to predict residue contacts and identify key amino acid combinations driving these interactions. By transforming contact prediction into a statistical testing problem for multivariate categorical data, CATParc accurately assesses the uncertainty of predictions without relying on pre-defined models. Could this framework unlock a deeper understanding of protein evolution and ultimately improve our ability to engineer proteins with desired properties?
The Emergent Logic of Proteins
The ability to forecast how alterations in a protein’s genetic code affect its behavior is fundamental to modern biological research and medical advancement. Proteins are the workhorses of cells, and even subtle changes at the molecular level can disrupt their function, leading to a vast array of diseases – from rare genetic disorders to common conditions like cancer and Alzheimer’s. Consequently, accurately predicting these functional consequences allows researchers to pinpoint the underlying causes of illness and identify potential therapeutic targets. Moreover, this predictive capability is driving the development of precision medicine, enabling the design of tailored treatments that address the specific molecular defects present in individual patients, and accelerating the creation of novel protein-based therapies with enhanced efficacy and reduced side effects.
Historically, determining how a mutation alters a protein’s function has proven remarkably difficult due to the intricate web of interactions within these molecules. Conventional computational approaches frequently simplify these relationships, treating proteins as static entities rather than the dynamic, flexible structures they truly are. This oversimplification often fails to capture the subtle, yet critical, effects of mutations on protein folding, stability, and interactions with other biomolecules. Consequently, predictions generated by these traditional methods often exhibit significant inaccuracies, hindering progress in fields like drug discovery and personalized medicine where precise understanding of protein behavior is paramount. The challenge lies not merely in identifying where a mutation occurs, but in deciphering how that change propagates through the protein’s complex architecture to ultimately impact its biological role.
Uncovering Hidden Relationships: A Sequence-Based Approach
CATParc is a model-free framework designed to analyze relationships between amino acids within protein sequences. Unlike methods relying on pre-defined structural models or energy functions, CATParc operates directly on sequence data, specifically Multiple Sequence Alignments (MSAs). This approach allows for the identification of co-evolving residue pairs without prior assumptions about protein structure or function. The framework’s core principle is to quantify the statistical dependence between amino acids, inferring potential functional or structural relationships based on observed patterns of variation across homologous sequences. By avoiding model-based constraints, CATParc aims to provide a more unbiased and comprehensive assessment of residue interactions.
Prior to statistical analysis, CATParc requires the conversion of raw Multiple Sequence Alignment (MSA) data into a numerical format suitable for computation. This is achieved through One-Hot Encoding, a process where each amino acid residue within the MSA is represented as a binary vector. The length of this vector corresponds to the total number of possible amino acids (typically 20 standard amino acids plus gap characters). A ‘1’ is placed at the index corresponding to the specific amino acid present at that position in the sequence, while all other indices are set to ‘0’. This transformation effectively converts qualitative sequence data into a quantitative format, allowing for the application of statistical methods like partial correlation analysis.
Partial correlation, as implemented within CATParc, assesses the linear relationship between two amino acid residues while controlling for the effects of all other residues in the multiple sequence alignment (MSA). This is achieved by calculating the correlation coefficient between residue pairs after removing the variance explained by intervening residues. Specifically, the partial correlation coefficient [latex]r_{xy \cdot z}[/latex] between residues x and y, conditioned on residue z, is derived from the covariance and variance matrices constructed from the MSA data. By accounting for these confounding factors, CATParc aims to identify direct, rather than indirect, relationships between residues, providing a more accurate representation of the structural and functional dependencies within the protein.
![CATParc consistently outperforms L2, SUP, and PSICOV in ROC analysis (represented by [latex]\square[/latex], [latex]\bigcirc[/latex], [latex]\bigtriangleup[/latex], and [latex]\lozengere[/latex] respectively), achieving higher power while maintaining control of the Type I error rate (indicated by the red dashed line at 0.05).](https://arxiv.org/html/2601.15566v1/x4.png)
Validating Accuracy: From Correlation to Confidence
CATParc employs a Spectrum-Based Test to assess the statistical significance of predicted residue contacts, moving beyond simple co-evolutionary analysis. This test calculates a p-value for each potential contact based on the observed frequency of correlated mutations across multiple sequence alignments. The spectrum considers the distribution of mutual information and assesses whether the observed correlation exceeds what would be expected by chance, accounting for factors like sequence depth and alignment quality. A stringent p-value threshold is then applied to filter for high-confidence residue contacts, minimizing false positive predictions and ensuring the reliability of the identified interactions.
CATParc achieves high accuracy in residue contact prediction, as quantitatively demonstrated by its performance across diverse protein families using the Area Under the Curve (AUC) metric. Evaluations have shown CATParc consistently outperforms existing methods in identifying residue pairs that are spatially close in the protein’s three-dimensional structure. Specifically, the AUC scores generated from testing CATParc on benchmark datasets indicate a statistically significant improvement in contact prediction capability compared to alternative computational approaches. This improved performance suggests a greater ability to accurately model protein structure and function through residue contact identification.
CATParc incorporates Group Lasso regularization to improve the selection of relevant variables during model training and increase the overall robustness of its predictions. This technique functions by grouping correlated variables and applying a penalty during model optimization, effectively shrinking the coefficients of entire groups to zero if they are not jointly predictive. Consequently, Group Lasso reduces the risk of identifying spurious residue contacts – minimizing Type I error rates – and provides more stable and interpretable results compared to methods lacking this regularization strategy. This approach is particularly beneficial when dealing with the high dimensionality and inherent correlations present in protein sequence data.
Decoding Functional Networks: Beyond Individual Residues
Proteins don’t function in isolation; rather, specific arrangements of amino acids work in concert to dictate a protein’s capabilities. CATParc excels at pinpointing these crucial amino acid combinations, moving beyond simple pairwise interactions to reveal cooperative effects. The framework assesses how changes in one amino acid influence another’s impact on protein function, thereby identifying synergistic or antagonistic relationships that would otherwise be missed. This detailed analysis goes beyond identifying important individual amino acids and uncovers the network of dependencies that define a protein’s operational mechanism, offering a more nuanced understanding of how sequence translates into function and providing a powerful tool for predicting the consequences of genetic mutations.
CATParc leverages the statistical method of Self-Normalized Partial Correlation to refine the analysis of amino acid interactions within proteins. Unlike traditional correlation methods susceptible to noise and spurious relationships, this technique effectively isolates the direct influence of each amino acid combination on protein function. By normalizing the partial correlations, the framework achieves greater stability and reduces the impact of confounding factors, ultimately leading to a more accurate depiction of functional dependencies. This approach proves particularly valuable when analyzing complex proteomes, as it allows researchers to confidently identify key amino acid pairings that drive protein behavior and distinguish them from incidental correlations, thereby enhancing the reliability of predictive models.
CATParc significantly enhances the precision of predicting how changes in a protein’s amino acid sequence affect its function. When combined with established Evolutionary Scale Modeling (ESM) techniques, the framework achieves a demonstrable 3% increase in prediction accuracy. This improvement, quantified by a Spearman correlation coefficient of 0.85, suggests a strong ability to discern the functional consequences of mutations. The enhanced predictive power has implications for understanding disease mechanisms, guiding protein engineering efforts, and ultimately, accelerating the development of novel therapeutics by more accurately assessing the impact of genetic variations.
The study demonstrates a compelling shift away from reliance on pre-defined models in understanding protein interactions. Rather than imposing hierarchical structures, CATParc allows patterns to emerge from the data itself, utilizing partial correlation to reveal relationships between residues. This approach aligns with the notion that complex systems benefit from encouraging local rules rather than centralized control. As Simone de Beauvoir observed, “One is not born, but rather becomes a woman.” Similarly, protein functionality isn’t predetermined, but emerges from the interplay of its constituent parts, a process CATParc effectively illuminates through its model-free inference of coevolutionary signals.
Beyond Prediction: Charting the Unseen
The framework detailed within prioritizes discerning interaction over imposing structure. CATParc, by focusing on partial correlation, reveals a network of influences rather than a predetermined architecture. This approach subtly shifts the focus: prediction, while useful, becomes secondary to understanding the emergent properties arising from amino acid co-evolution. The real challenge lies not in simply anticipating contact, but in characterizing the conditions under which that contact becomes functionally significant – or doesn’t. The precision matrix offers a glimpse, but its interpretation demands acknowledgement that correlation does not equate to causation; it highlights relationships, not commands.
Future work should address the limitations inherent in inferring function solely from static structural data. Proteins exist in dynamic states, and the influence of environmental factors, post-translational modifications, and allosteric regulation remain largely unexplored within this statistical lens. Perhaps the most potent extension would involve integrating time-series data, allowing for the characterization of how co-evolutionary patterns change in response to external stimuli.
Ultimately, the field progresses not by seeking complete control over protein behavior, but by refining the tools for observing and interpreting its intrinsic order. Sometimes, the most valuable insight comes from recognizing where prediction fails, and acknowledging that inaction-allowing the system to reveal itself-can be the most effective strategy.
Original article: https://arxiv.org/pdf/2601.15566.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
2026-01-25 03:28